183-又学了点GSEA的新知识
刘小泽写于2020.4.29 GSEA的知识之前学习过: 富集分析Enrich me again! Pathway通路与GSEA基因集有何区别? 豆豆学习GSEA GSEA–从基因到美图 这次来介绍点新知识
前言
如果我们感兴趣某个基因,想看看它在什么通路中发挥作用。例如:DDIT3,就可以在https://www.kegg.jp/kegg/pathway.html中选择Organism为“hsa”,然后输入基因名,看到这个基因参与了6个通路,其中可能当前研究比较感兴趣这个细胞凋亡通路: hsa04210

然后想对这个通路做个GSEA,看看我们的基因是在这个通路上调还是下调
GSEA分析的过程
重点需要两个因素:一个是基因集GeneSet(也就是这里的hsa04210中包含的基因),另一个是我们自己的基因列表GeneList
需要注意的是:基因列表GeneList需要将基因按照logFC从大到小排序
logFC的FC即Fold Change,是treat/control。如果logFC>0(即FC>1),表示基因在treat组上调;反之下调 常规的差异分析需要人工设置阈值,比如logFC>1 & padj < 0.05来确定上下调差异基因,之后再进行富集分析 但是GSEA不同,例如我们总共得到了15000个差异基因,经过筛选蛋白编码基因和基因ID转换以后,最后剩余12000个。我们就是利用这12000个基因降序排列后的logFC值进行的GSEA分析
结合GSEA的文章来看:https://www.pnas.org/content/102/43/15545
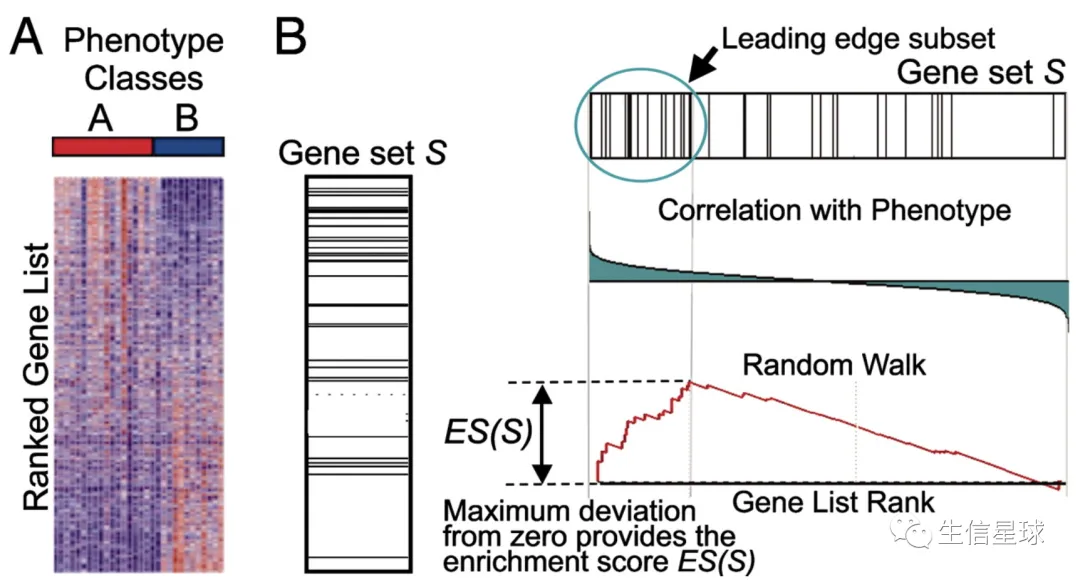
图A,也就是A组(treat)和B组(control)之间的差异基因,按照logFC从大到小排列【A是红色B是蓝色表示treat中上调,颜色越深表示logFC绝对值越大】
图B,就是展示了计算过程,我们把选好的GeneSet(例如hsa04210中包含的基因)去沿着我们排序好的GeneList走一遍:
看到的折线就是:
- The score is calculated by walking down the list L(GeneList), increasing a running-sum statistic when we encounter a gene in S(GeneSet) and decreasing it when we encounter genes not in S.
- The magnitude of the increment depends on the correlation of the gene with the phenotype.
代码部分直接看花花之前发的: GSEA–从基因到美图
对结果的思考
运行结束后,会返回一个gseaResult 对象,把它变成数据框后就能清楚看到富集到的通路

但这个setSize什么意思呢?最后一列的core_enrichment又是什么意思?
来看这个setSize
首先会想,它是不是指这个通路的基因数量?这个很好验证
以hsa04210为例,可以在网页中看到这个通路包含的基因数量

或者在R中:length(kk@geneSets$hsa04210) 【这个kk就是利用clusterProfiler运行GSEA的结果】
看到:这个通路总共136个基因
所以,并不是代表这个通路所有的基因
它其实代表了:GeneSet在GeneList中存在的基因
setsize=kk@geneSets$hsa04210[kk@geneSets$hsa04210%in%names(geneList)]
length(setsize)
# 116个
也就是说:只有116个hsa04210通路基因存在于我们的GeneList中
【再看👆右上角的图,其中中每一条线表示一个基因,而且这个基因是在hsa04210通路和按logFC排序后的geneList中存在的基因】
最后一列的core_enrichment
这个看看这一列的内容,然后猜测一下,应该是某些基因名。但是哪些基因呢?是上面👆这个通路和geneList的交集(116个基因)还是?
如果是全部的,就不应该叫core... ,既然列为了“核心”基因,那么就应该是贡献较大的基因们。想必都看过GSEA的结果图,是有峰的,然后结合前面说的GSEA计算方法,那么猜测:应该是指导致峰出现的这部分基因
在官网就有解释:https://www.gsea-msigdb.org/gsea/doc/GSEAUserGuideFrame.html (下面略作修改) Genes that contribute to the leading-edge subset within the gene set. This is the subset of genes that contributes most to the enrichment result.
把它们提取出来:
# kk是gseKEGG运行的结果
y=as.data.frame(kk)
core_g=str_split(y['hsa04210',]$core_enrichment,"/",simplify = T)
tmp=bitr(core_g,
fromType = "ENTREZID",
toType = "SYMBOL",
OrgDb = org.Hs.eg.db)
tmp2=merge(tmp3, new_list, by = "SYMBOL")[c(1,4,2,3)]
# 按logFC降序排列,和GSEA图上的线条吻合
tmp2=tmp4[order(tmp4$logFC,decreasing = T),]
之后得到这个基因列表(就是上图👆中绿色圆圈的基因们),就可以看看这些核心的上调基因【因为峰出现在头部,所以这些核心基因都是上调的;如果峰出现在尾部,那么核心基因就是下调的】的作用是促进还是抑制细胞凋亡。如果发现这部分上调基因的大部分是促进凋亡,那么处理组在经过处理后更可能产生了促凋亡的影响