167-IGV载入bam后出错怎么办?
刘小泽写于2020.2.16 虽然是一个小问题,但也不能慌
关于IGV的介绍
之前写过几篇:
- 必备可视化Integrative Genomic Viewer(IGV)(一)
- 必备可视化Integrative Genomic Viewer(IGV)(二)
- 2019意犹未尽的基因组可视化IGV(一)
- 2019意犹未尽的基因组可视化IGV(二)
- 勇敢跳进igv安装的坑
- IGV加载很久很烦人?三步帮你解决!
目前,官网更新到了2.8.x版本
问题来源
先做一个测试的小bam文件
先将bam文件进行sort
samtools sort -@ 4 -o test.sort.bam test.bam
然后只要它的1号染色体
samtools view -h test.sort.bam chr1 | samtools view -Sb - > test.small.bam
最后构建索引
samtools index test.small.bam
把bam同.bai一起导出来
将bam放入IGV中,一开始是没有问题的
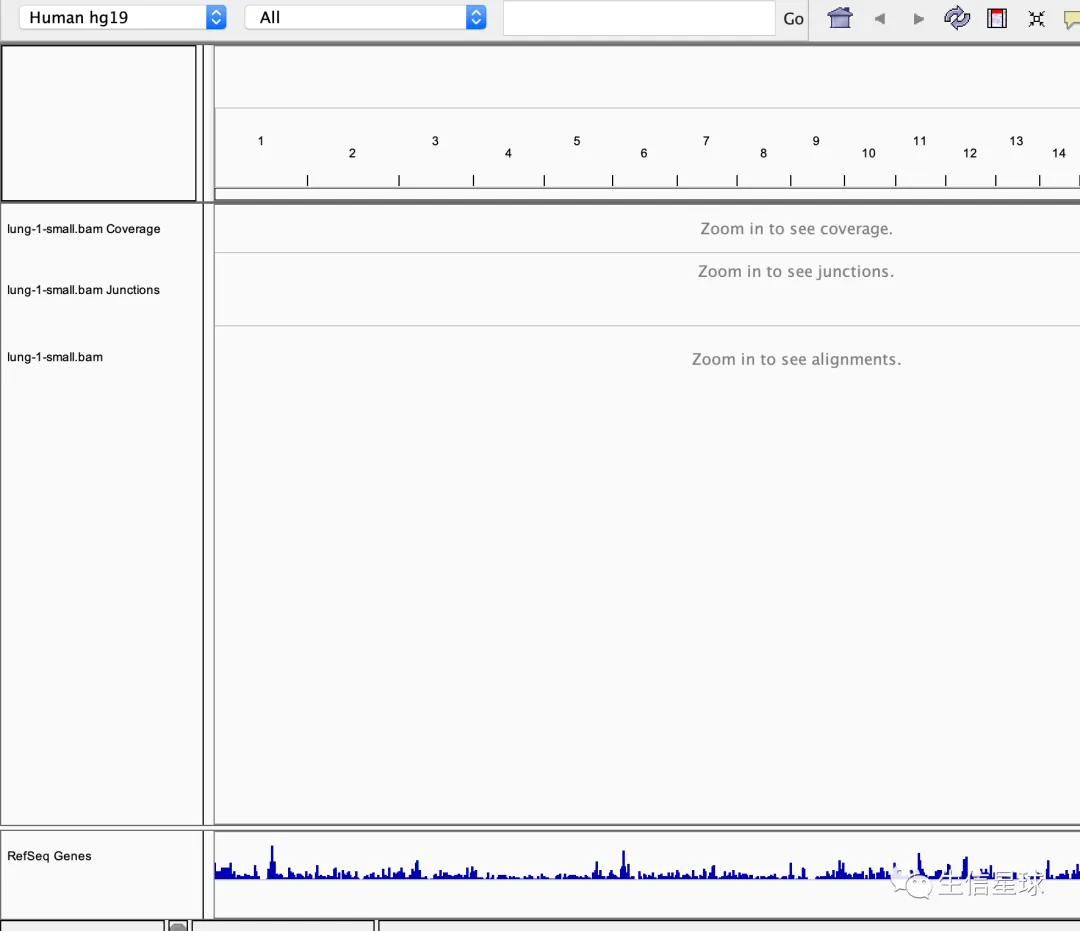
但当放大想查看具体的比对结果时,出现了:

应急方法
这其实就是索引文件的问题,怎么办?不要惊慌,其实也不需要去服务器重新做一遍索引、再导出,我们直接利用IGV自带的igvtools工具就好了,先能完成任务即可
在Tools =》 Run igvtools , 然后选择Index ,并且指定好文件的位置
关于这个位置信息:不要手动输入,这样可能会输错。
windows利用我的电脑中显示的的文件链接:
mac可以直接把文件拖进terminal,路径会立刻出来:

然后运行即可:
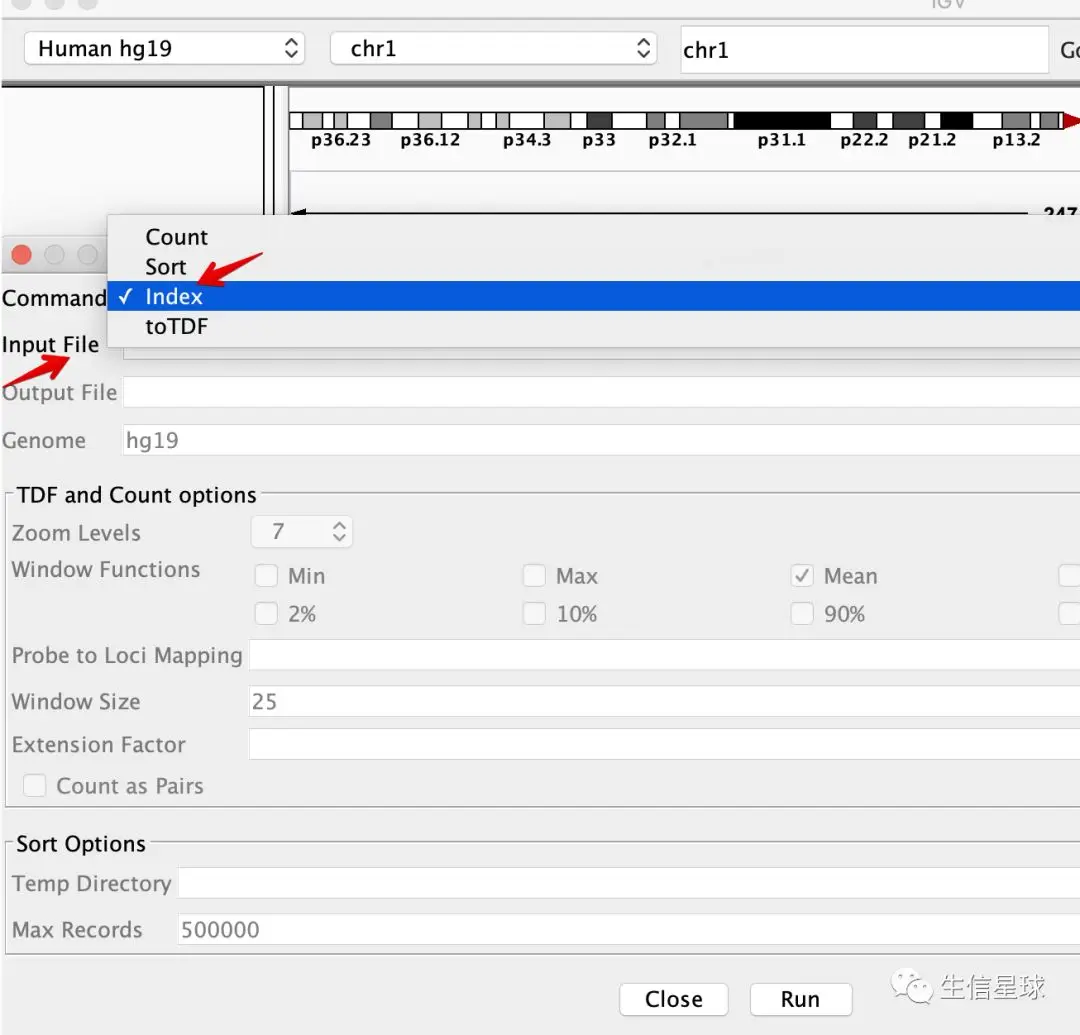
之后利用它构建的index,bam文件就能顺利加载进来啦