158-刘小泽学习GSEA
刘小泽写于2020.1.22-23 好久没有更新了,赶快更新一波 我们经常听说:Gene Set Enrichment Analysis、GSEA、基因集富集分析,这三种实际上都是描述的一种方法,就是寻找基因的功能大概是什么样子的。 这一次,就来认识一下GSEA吧
以往的认知
我们经常做转录组分析,于是最熟悉的是:差异分析 + 功能注释。经常设定一个logFC阈值,然后找变化倍数大于或者小于这个值的基因,作为上调或者下调。而这个阈值的设定,经常没有一个标准,主观性很大,但有时也会添加一点统计知识进去,比如利用mean + 2sd来计算这个阈值。
最后会利用这些差异基因,来进行GO或者KEGG的注释,找到它们主要在哪些通路富集,从而提供一些思路。
那么以上👆的思路就属于:过表征分析(ORA, Over-Representation Analysis),属于初步探索。
当然其中也会有一些问题,例如:
- 只分析差异基因,但表达真的只受那些变化大的基因影响吗?
- 我们设置的logFC靠谱吗?不同的logFC得到的差异基因数量不同,那么再进行富集分析的结果又有多少重合呢?
- 找差异基因的过程往往假设基因间是相互独立的,但实际上许多基因的关系还是很密切的
- 另外即使两个基因都被分在了上调基因组,一个FC是8,一个FC是4,那么能认为这两个基因的特征是一样的吗?
- 如果现在有两个实验组,都分别找到了差异基因,现在想通过找它们共同的差异基因,然后做富集分析。但是,结果可能是:两个组的差异基因数量本身很多,它们的交集很少(比如才二三十个),这样再向下做差异分析就很困难
之前也介绍过相关的内容,见: 富集分析Enrich Me! 、 富集分析Enrich me again!
GSEA就是属于第二代方法:FCS(Functional Class Scoring)的范畴
基本上能看到类似的这种图就说明是用GSEA做的:
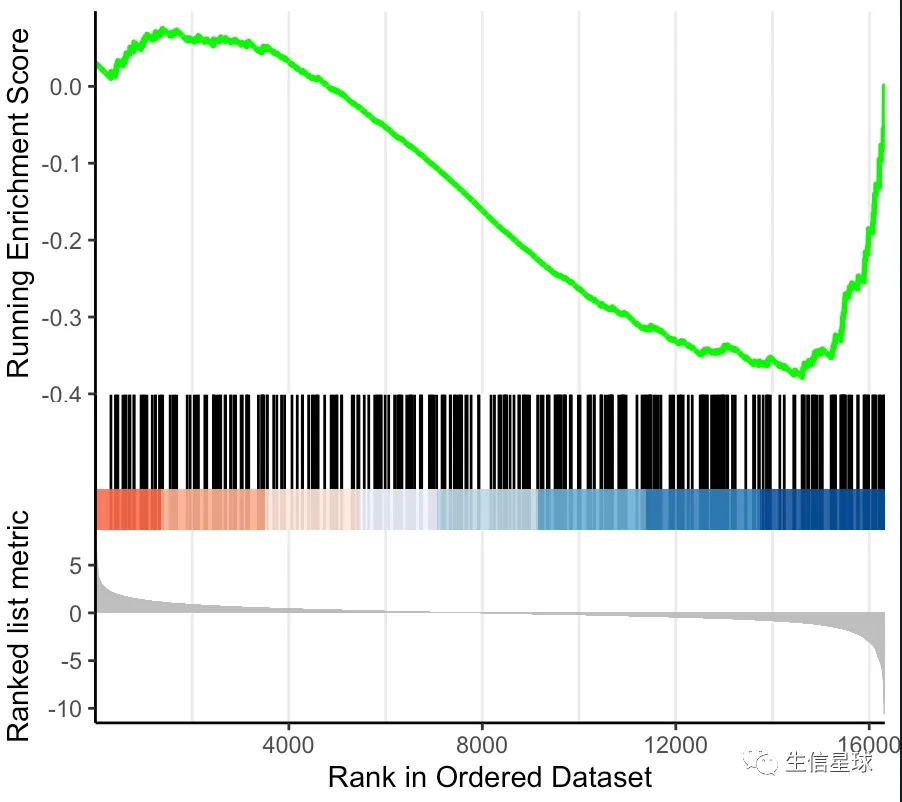
那么怎么认识这个GSEA呢?
毕竟之前和它大的交道少,于是需要大体了解一下(但不是说去钻研算法)
从原理开始
- 和差异分析类似,GSEA也是需要计算treat-control组的差异(比如log2FC),但是它先不设定阈值
- 然后从大到小对差异倍数排序,并且知道这个排序结果对应的基因名=》也就是得到了一个基因列表(L)
- 从计算方法就能得知,这个基因列表的一侧主要是treat的特征,另一侧主要是control的特征
- 之后,使用已经发布并且公认的基因集做检验,这个基因集主要是从MSigDb(分子标签数据库 http://software.broadinstitute.org/gsea/msigdb/index.jsp)获得,其中会包括GO、KEGG、Reactome等注释内容
其实排好序之后,我们可以手动去基因列表L的两侧去查,看看有没有我们想要的基因,但往往一个基因列表动辄上万,因此这个方法虽然可以做,但软件和统计知识并不允许我们去这么干。
于是,事先将已知的通路(包含相关的基因信息)存储起来,用的时候直接去验证它们就好了,而这个就是基因集富集分析的基因集。比如要看某个GO term在我们排序好的基因列表中富集在头部还是尾部,就能反证我们的基因集中treat组上调或者下调基因是否属于这个通路
**GSEA方法的原假设是:**某个通路的全部基因在我们排序后的基因列表中随机分布,如果我们看到它们”意外“出现在基因列表的某一端(从图上看是在某一侧形成一个峰),那么就可以计算显著性来看看富集程度如何。如果富集结果显著,那么就拒绝原假设,认为这个通路的基因在我们的基因列表中富集,并且看到富集分数
名词解释
富集分数:这个名词是个重点,划下来! 英文名称是:ES(Enrichment score)。当使用基因集的基因来遍历我们的基因列表时,就会判断基因集的基因是不是存在于基因列表。如果存在就加分,不存在就减分。但具体的分数计算规则看看这个图就好(https://bioinformatics.cancer.gov/sites/default/files/course_material/GSEA_Theory.pptx)。只要明白GSEA的那条曲曲折折的线就是通过不断的加分减分做出来的就行了。那些对ES值有贡献的基因称为 Leading Edge Subset
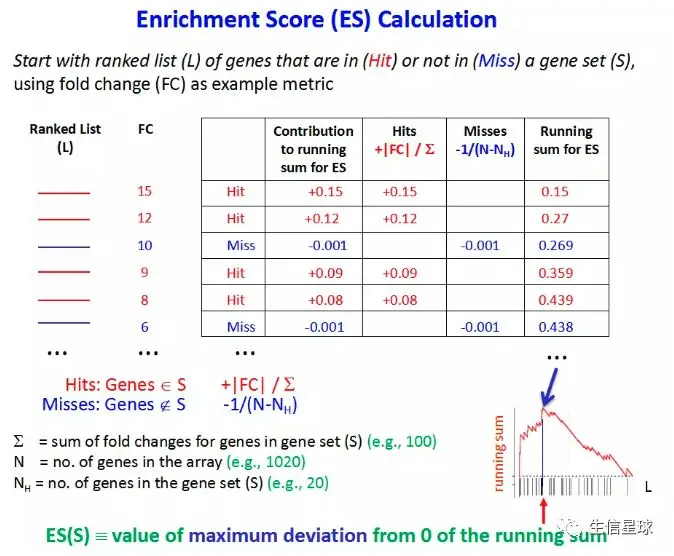
基因列表的排序规则:前面提到我们会对差异分析结果使用
log2FC进行排序,当然这只是GSEA的排序规则中的一种。它的默认方法是:Signal to Noise Ratio。一般对于treat-control这种分组的样本,可以使用:Signal2Noise,t-test,Ratio of Classes【也就是fold change】,log2_Ratio_of_Classes【也就是log2FC】;另外,还有一种连续型的样本(例如时间序列样本),它们可以用:Pearson,Cosine,Manhattan,Euclidean方法【详细看:http://software.broadinstitute.org/gsea/doc/GSEAUserGuideTEXT.htm#_Metrics_for_Ranking】
参考资料
GSEA怎么分析
方法一:R语言
首先也是做一个差异分析,可以用limma、edgeR或者DESeq2
然后需要一个基因列表,按照log2FC从大到小排序
# 得到差异分析结果:DEG_mtx
geneList <- DEG_mtx$log2FoldChange
# 如果是Ensemble ID,并且如果还带着版本号,需要去除版本号,再进行基因ID转换,得到Entrez ID
names(geneList) <- str_split(rownames(DEG_mtx) ,
pattern = '\\.',simplify = T)[,1]
geneList_tr <- bitr(names(geneList),
fromType = "ENSEMBL",
toType = c("ENTREZID","SYMBOL"),
OrgDb = org.Hs.eg.db)
new_list <- data.frame(ENSEMBL=names(geneList),
logFC = as.numeric(geneList))
new_list <- merge(new_list, geneList_tr, by = "ENSEMBL")
geneList <- new_list$logFC
names(geneList) <- geneList_tr$ENTREZID
# 最后从大到小排序,得到一个字符串
geneList <- sort(geneList,decreasing = T)
再利用clusterProfiler进行GSEA分析
如果进行GO分析
# 想看biological process(BP),可以先不过滤p值 go_result <- gseGO(geneList = geneList, ont = "BP", OrgDb = org.Hs.eg.db, minGSSize = 10, maxGSSize = 500, pvalueCutoff = 1) # 将其中的基因名变成symbol ID go_result <- setReadable(go_result, OrgDb = org.Hs.eg.db) # 还可以直接点击查看,只需要转成数据框 go_result_df <- as.data.frame(go_result)如果对结果的某个通路感兴趣,可以作图
gseaplot2(go_result, geneSetID = c("GO:0006695"))如果要进行KEGG分析
kk <- gseKEGG(geneList = geneList, organism = 'hsa', nPerm = 1000, minGSSize = 10, maxGSSize = 500, pvalueCutoff = 1, verbose = FALSE)作图依然是:
gseaplot(kk, geneSetID = "hsa04110")
方法二:GSEA软件
搞定输入
软件需要三个输入文件,分别是:
表达谱文件(.gct文件或者符合要求的.txt)。GSEA原来提供了RNA-seq原始数据的标准化、差异分析等一系列流程,也就是说提供原始count矩阵是可以的。但现在,官方建议先使用其他方法(例如Deseq、limma等)进行标准化后,再导入

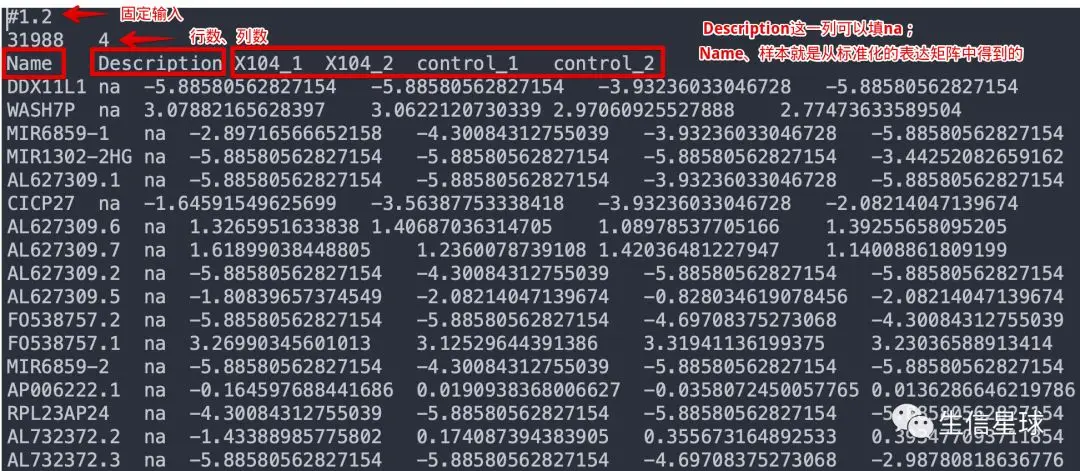
样本信息文件(.cls)
cat(c(4,2,1,"\n"),file = "test.cls",sep = " ",append = F) cat(c("#trt","control","\n"),file = "test.cls",sep = " ",append = T) cat(c(rep(c("trt","control"),each=2),"\n"),file = "test.cls",sep = " ",append = T)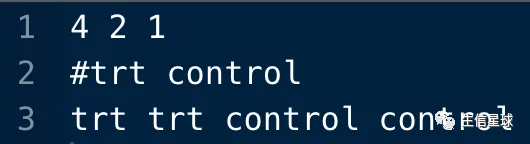
基因集文件(.gmt),这个可以从https://software.broadinstitute.org/gsea/downloads.jsp#msigdb这里下载,也可以在软件中直接加载
之前技能树写过:https://www.jianshu.com/p/b409a5576ce1 其中结合芯片数据介绍了:批量运行GSEA,命令行版本;制作自己的gene set文件给gsea软件
调整参数
参考:http://software.broadinstitute.org/gsea/doc/GSEAUserGuideTEXT.htm#_Run_GSEA_Page
Numbers of permutation:当分析多个基因集时,就要注意这个多重假设检验。就相当于多次抽样,次数越多越准确。这个默认是1000次。但建议先从小的数字开始尝试运行(例如10),先完成再完美
Permutation type:这里有两个选择,一个是phenotype,一个是gene_set。第一个phenotype效果最好,官方建议如果每个组的样本都在7个以上,就使用phenotype;反之如果有一个组少于7个样本,那么就用gene_set【另外,在GSEA 4.0之前的版本,gene_set叫做tag permutation】
Phenotype Labels:一般是设置
treat vs control【参考:http://software.broadinstitute.org/gsea/doc/GSEAUserGuideTEXT.htm#Phenotypes】
查看结果
程序一般会在主目录下新建一个目录:gsea_home,然后 output 里面按照日期进行排序

里面的文件会非常多,但有一个总览index.html:
就会看到:
- 第一个结果是处理后上调基因倾向于富集的通路
- 第二个结果是处理后下调基因倾向于富集的通路
并且它们都可以直接点开看结果,或者将excel读取到R语言中进行处理

如果要把结果读取到R中
结果文件也是存放在了index.html同级目录中,就是一个excel表格
GSEA_result_up <- readr::read_delim("gsea_report_for_trt_9111157956735.xls",delim = "\t")
GSEA_result_up <- GSEA_result_up %>%
select(NAME,NES,SIZE,`FDR q-val`)
