152-ID转换失败,爬起来继续high
刘小泽写于19.12.9 昨天遇到一个基因ID转换的问题,才发现原来ID转换不是这么简单…
0 前言
拿到一个接近60000 x 6000的原始表达矩阵,行是基因,列是样本。并且行名都是Ensembl ID,还带着版本号。
我的第一直觉就是先把ID转换成Symbol,保证后续操作的易读性
下面👇是我对这个原始矩阵的操作
1 第一步 对原始表达矩阵进行过滤
很简单粗暴,直接去掉那些在所有样本不表达的基因
> dim(origin_count)
[1] 58347 6000
# 过滤后的矩阵df
df <- origin_count[rowSums(origin_count)>0,]
> dim(df)
[1] 43523 6295
2 第二步 修改原始行名的Ensembl ID
这里要先说一下Ensembl 的ID
2.1 我们常见的Ensembl ID比如:
ENSG00000279928.1 ENSG00000279457.2 详细说明:https://asia.ensembl.org/Help/Faq?id=488
- 其中,ENS是固定字符,表示它是属于Ensembl数据库的。默认物种是人,如果是小鼠就要用
ENSMUS开头,关于物种代码:http://www.ensembl.org/info/genome/stable_ids/index.html - G表示:这个ID指向一个基因;E指向Exon;FM指向 protein family;GT指向gene tree;P指向protein;R指向regulary feature;T指向transcript
- 后面11位数字部分如
00000279928表示基因真正的编号 - 小数点后不一定每个都有,表示这个ID在数据库中做了几次变更,比如
.1做了1次变更,在分析时需要去除
至于怎么去掉小数点后面的信息也很简单,之前花花介绍过( 去掉ensembl id的version信息)
2.2 小心两个坑
🤡 第一坑
不过这里,仅仅是简单去掉还不够,因为我们的目的是让新的Ensembl ID当做行名,但是去掉小数点后会出现一个问题:重复值 ,而行名是不允许重复值出现的
因此,需要保证新的Ensembl ID中如果出现重复值,那么就只保留第一个
🤡 第二坑
之前原始的Ensembl ID是和行名的长度一致的,现在得到了去重复的Ensembl ID,那么由于长度不等,因此不能一一对应到行名上。
要根据之前Ensembl ID的重复情况,去掉原始数据框对应的行(不过不要担心,重复的值没有多少,所以也去不掉多少行,对结果没有太大影响;并且既然是重复的行,它们的表达量应该也是类似的,所以取其中一个即可)
2.3 那么整体的操作如下:
head(rownames(df),3)
# "ENSG00000000003.14" "ENSG00000000005.5" "ENSG00000000419.12"
# 去掉小数点后的gene name(gn)
gn <- stringr::str_split(rownames(df),"\\.",simplify = T)[,1]
# 根据gn的重复情况,df也进行对应的去除
df <- df[!duplicated(gn),]
# 此时df的行数才和gn相等
rownames(df) <- gn[!duplicated(gn)]
> dim(df) # 看到相比之前(43523行),过滤掉了39个重复行
[1] 43484 6295
> head(rownames(df),3)
[1] "ENSG00000000003" "ENSG00000000005" "ENSG00000000419"
3 第三步 基因ID转换
3.1 使用org.Hs.eg.db + mapIds
suppressMessages(library(org.Hs.eg.db))
symb <- mapIds(org.Hs.eg.db, keys=rownames(df),
keytype="ENSEMBL", column="SYMBOL")
> length(rownames(df))
[1] 43484
> length(na.omit(symb))
[1] 22965
可以看到基本包含了一半的NA值
3.2 使用org.Hs.eg.db + bitr
其实结果一样,因为都是使用的org.Hs.eg.db数据库,这里只是列一下方法
library(clusterProfiler)
library(org.Hs.eg.db)
gene_tr <- bitr(rownames(df), fromType = "ENSEMBL",
toType = c("SYMBOL"),
OrgDb = org.Hs.eg.db)
# 47.19% of input gene IDs are fail to map...
> length(na.omit(symb_2$SYMBOL))
[1] 23196
可以看到比mapIds能多得到一些基因
疑问
**这么多的NA基因,难道真的不存在对应的Symbol基因名吗?**这个问题值得思考🤔
很多时候,使用了两种工具,得到差不多的结果,可能就认为事已至此了。但是换种角度思考问题,如果问题不是出在工具呢?如果是数据库呢?
好,接下来就看看没有对应Symbol的那些Ensembl ID
not_mapped_ensembl <- names(symb[is.na(symb)])
> length(not_mapped_ensembl)
[1] 20519
> head(not_mapped_ensembl,3)
[1] "ENSG00000002079" "ENSG00000018607" "ENSG00000020219"
那么随便挑一个(例如第一个)直接复制、搜索,看看到底有没有基因名
搜索的结果让人大吃一惊,竟然它真的有基因名,而且还在HGNC、Ensembl、Genecards等数据库有收录
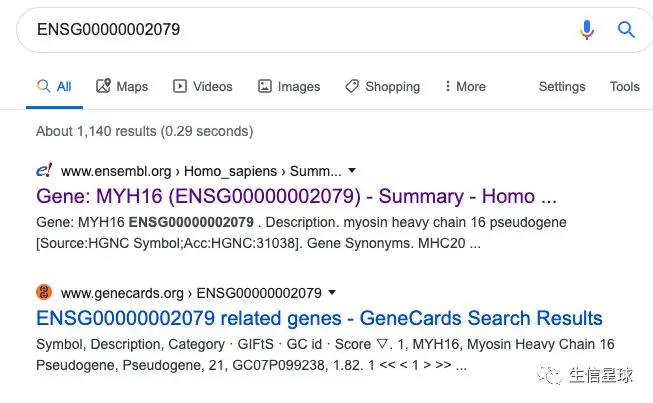
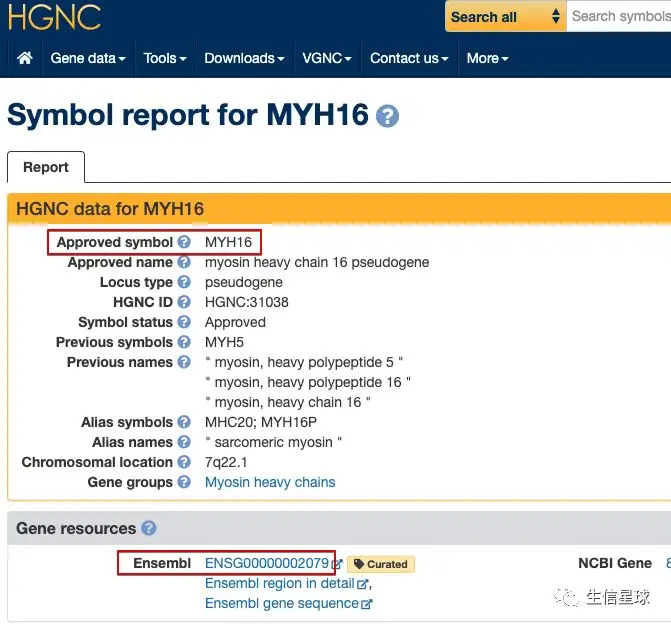
看到Ensembl 列出的该基因的信息很全,不由得想到,基因ID转换不止这两种工具,还有一种:Ensembl自家的biomaRt,尝试一下
3.3 使用hsapiens_gene_ensembl + biomaRt
看它的帮助文档就很清楚它怎么操作,过程也很简单
library("biomaRt")
# 用useMart函数链接到人类的数据库
ensembl <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")
# 除以以外还有很多:使用 listDatasets(ensembl) 查看
# 依然是要列出查询的gene ID,以及要转换的格式
# 【biomaRt强大之处就在于它的转换格式种类非常之丰富】
attributes <- listAttributes(ensembl)
View(attributes) # 查看转换格式
# 列出要转换的ID(比如前面匹配失败的3个基因)
value <- head(not_mapped_ensembl,3)
# 列出当前格式和要比对的格式(当前是ensembl_gene_id;我要比对hgnc_symbol和chromosome_name)
attr <- c("ensembl_gene_id","hgnc_symbol","chromosome_name")
# 最后运行就好了(其中filters指定当前基因 ID类型)
ids <- getBM(attributes = attr,
filters = "ensembl_gene_id",
values = value,
mart = ensembl)
最后看一下结果,而且这个结果扩展性极强:
> ids
ensembl_gene_id hgnc_symbol chromosome_name
1 ENSG00000002079 MYH16 7
2 ENSG00000018607 ZNF806 2
3 ENSG00000020219 CCT8L1P 7
biomaRt的小问题
当提供的基因数量太多时,容易断线;它的检索速度非常慢!【不过用来应急还是可以的】
心得
当使用某个或某几个工具得到的结果感觉不满意时,不要只考虑工具的优劣,还要思考它们在后台帮我们做了什么。
比如这里基因ID的转换,靠的其实就是背后的数据库,工具虽有差异但没那么大(比如bitr比mapIds多了几个基因)。有时一条路走不通,换个工具“背后的”数据库未尝不是个好的解决办法!