140-利用ChIPpeakAnno进行下游分析
刘小泽写于19.10.11 是时候学习ChIP-seq的分析了,这次跟着Bioconductor的教程走一下ChIPpeakAnno的基本流程
前言
官网教程在:https://www.bioconductor.org/packages/devel/bioc/vignettes/ChIPpeakAnno/inst/doc/pipeline.html
更新于2019-7-26
这次先进行第一部分,将会进行以下操作:
将BED/GFF文件转为GRanges、两个peak数据集中寻找overlap、利用韦恩图可视化共有和特异的peaks
1 导入BED和GFF数据并获得overlap peaks
ChIPpeakAnno包需要从ChIP-seq实验等获得的peaks在基因组染色体的坐标信息,这些信息可以存放在BED、GFF、MACS、GRanges中,这里将会利用GRanges进行后续分析。而像BED, GFF 和MACS转为GRanges也是很容易的,利用toGRanges函数即可
下面👇的例子中,就将BED和GFF转为了GRanges,另外将原来peaks的注释信息通过addMetadata重新添加到GRanges中【这些文件都是包自带的,加载包后直接使用】
这个包需要依赖几十个R包
# 查看包示例数据所在位置
> system.file("extdata",package="ChIPpeakAnno")
[1] "/Library/Frameworks/R.framework/Versions/3.5/Resources/library/ChIPpeakAnno/extdata"
可以看到其中的MACS_output.bed文件是这样的
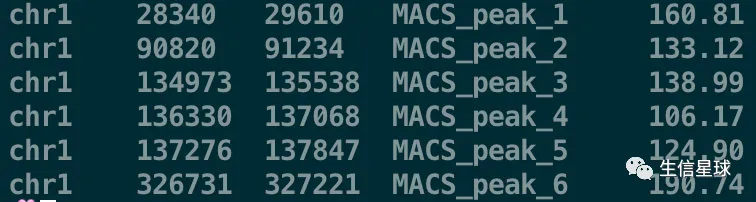
将BED文件转换为GRanges对象
bed <- system.file("extdata", "MACS_output.bed", package="ChIPpeakAnno")
gr1 <- toGRanges(bed, format="BED", header=FALSE)
> gr1
GRanges object with 230 ranges and 1 metadata column:
seqnames ranges strand | score
<Rle> <IRanges> <Rle> | <numeric>
MACS_peak_1 chr1 28341-29610 * | 160.81
MACS_peak_2 chr1 90821-91234 * | 133.12
MACS_peak_3 chr1 134974-135538 * | 138.99
MACS_peak_4 chr1 136331-137068 * | 106.17
MACS_peak_5 chr1 137277-137847 * | 124.9
... ... ... ... . ...
对gff文件转换
gff <- system.file("extdata", "GFF_peaks.gff", package="ChIPpeakAnno")
gr2 <- toGRanges(gff, format="GFF", header=FALSE, skip=3)
下面找bed和gff的重叠peaks
需要注意的是,使用findOverlapsOfPeaks函数需要将二者的格式调成一致。当前一个是数值型一个是整型
> class(gr1$score)
[1] "numeric"
> class(gr2$score)
[1] "integer"
先将gr2的转换一下格式,然后找overlap
gr2$score <- as.numeric(gr2$score)
ol <- findOverlapsOfPeaks(gr1, gr2)
找到以后添加metadata,例如添加
ol <- addMetadata(ol, colNames="score", FUN=mean)
这个ol是一个列表,我们可以看看其中包含的peaks信息,例如:
# gr1和gr2都有的peaks数量
> length(ol$peaklist$`gr1///gr2`)
[1] 166
# 仅gr2有的peaks数量
> length(ol$peaklist$gr2)
[1] 61
# 仅gr1有的peaks数量
> length(ol$peaklist$gr1)
[1] 62
要查看重叠的这些peaks信息的话(以前两行为例):
ol$peaklist[["gr1///gr2"]][1:2]
# 或 ol$peaklist$`gr1///gr2`
## GRanges object with 2 ranges and 2 metadata columns:
## seqnames ranges strand | peakNames
## <Rle> <IRanges> <Rle> | <CharacterList>
## [1] chr1 713791-715578 * | gr1__MACS_peak_13,gr2__001,gr2__002
## [2] chr1 724851-727191 * | gr2__003,gr1__MACS_peak_14
## score
## <numeric>
## [1] 850.203333333333
## [2] 29.17
## -------
## seqinfo: 1 sequence from an unspecified genome; no seqlengths
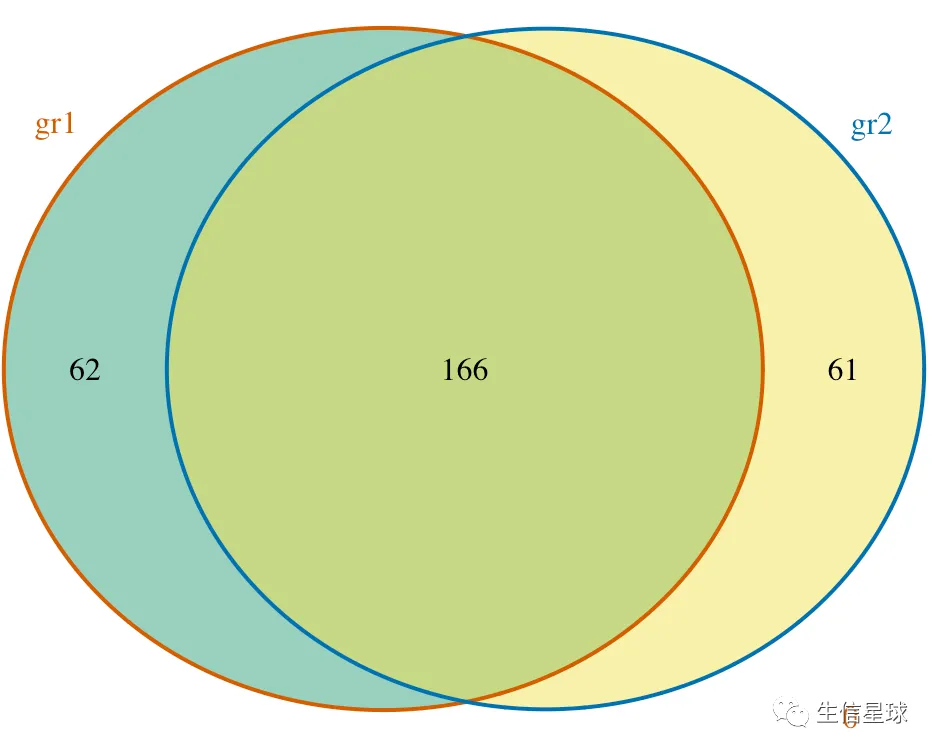
另外它还做了一个韦恩图使用的数据
> ol$venn_cnt
gr1 gr2 Counts
[1,] 0 0 0
[2,] 0 1 61
[3,] 1 0 62
[4,] 1 1 166
attr(,"class")
[1] "VennCounts"
做出韦恩图
makeVennDiagram(ol, fill=c("#009E73", "#F0E442"), # circle fill color
col=c("#D55E00", "#0072B2"), #circle border color
cat.col=c("#D55E00", "#0072B2")) # label color, keep same as circle border colors
2 准备注释数据
注释数据和peaks一样,也要是GRanges对象,这种注释不仅可以使用BED、GFF等文件,还能用EnsDb、TxDb等对象,使用toGRanges 进行创建
# 接下来就是利用EnsDb创建注释
library(EnsDb.Hsapiens.v75) ##(hg19)
annoData <- toGRanges(EnsDb.Hsapiens.v75, feature="gene")
> annoData[1:2]
GRanges object with 2 ranges and 1 metadata column:
seqnames ranges strand | gene_name
<Rle> <IRanges> <Rle> | <character>
ENSG00000223972 chr1 11869-14412 + | DDX11L1
ENSG00000227232 chr1 14363-29806 - | WASH7P
-------
seqinfo: 273 sequences from GRCh37 genome
3 结合位点可视化
binOverFeature()
找到重叠的peaks之后,就可以用binOverFeature来对基因组feature(例如转录起始位点TSS)附近的重叠peaks进行可视化
# 需要两个东西:重叠的peaks、注释信息annoData
overlaps <- ol$peaklist[["gr1///gr2"]]
binOverFeature(overlaps, annotationData=annoData,
radius=5000, nbins=20, FUN=length, errFun=0,
ylab="count",
main="Distribution of aggregated peak numbers around TSS")
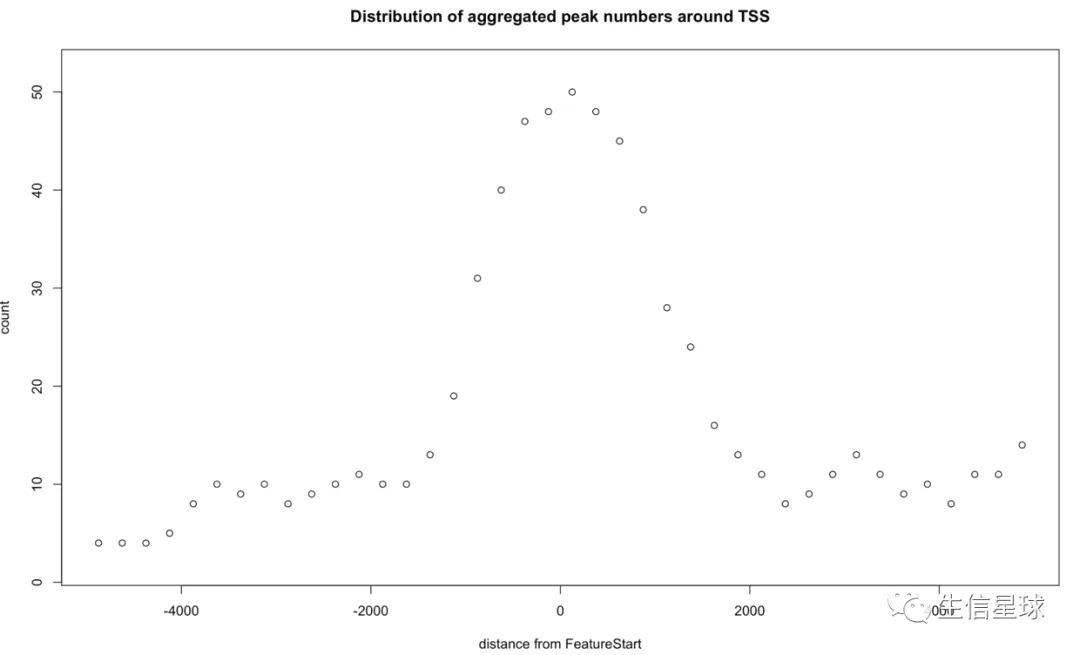
问:从这个图中能看出什么?
assignChromosomeRegion()
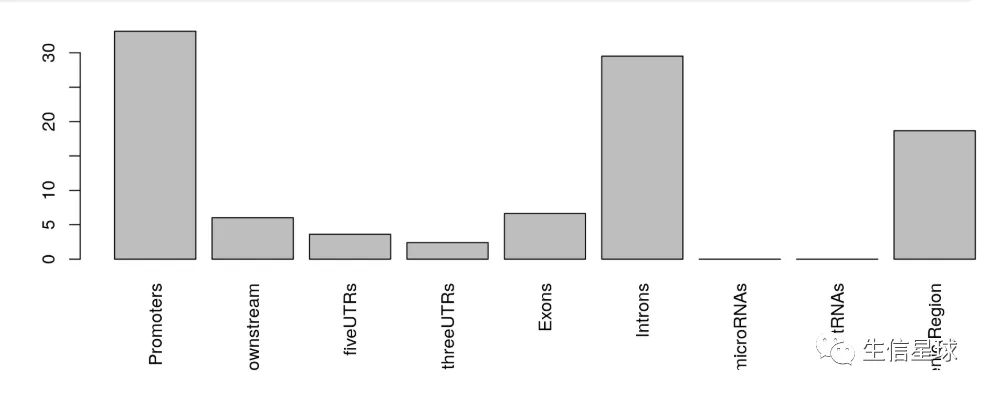
这个函数可以对peaks的分布区域进行概括,包括外显子、内含子、增强子、上游启动子/近端启动子、5’ UTR 、3’ UTR。可能一个peak会跨越多种类型的基因feature信息,因此可能看到输出结果中注释到的feature数量比输入的peaks数量更多。
peaks的分布有两种统计方式:peak centric or nucleotide centric ,默认使用前者
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
aCR<-assignChromosomeRegion(overlaps, nucleotideLevel=FALSE,
precedence=c("Promoters", "immediateDownstream",
"fiveUTRs", "threeUTRs",
"Exons", "Introns"),
TxDb=TxDb.Hsapiens.UCSC.hg19.knownGene)
barplot(aCR$percentage, las=3)