016-Bigbang~BLAST
刘小泽写于18.7.26
身处信息大爆炸时代,无限信息波涛汹涌,如何去繁存简? 活跃生物多学科领域,海量测序数据更迭,怎样千里挑一?
一声巨响,NCBI为生命科学带来了BLAST
背景知识
发展进程:
上世纪80年代中后期,核酸、蛋白质序列与结构的数据库中积累了大量的数据。1988 年 11 月,位于美国首都华盛顿北郊的美国国家生物技术信息中心(National Center for Biotechnology Information,简称 NCBI)成立。它隶属美国国家医学图书馆(National Library of Medicine,简称 NLM),而 NLM 又是美国国家健康研究院(National Institutes of Health,简称 NIH)的一个分支机构。NCBI 建立初期,仅 8 名人员,经过近 30 年的建设,NCBI 已发展成**国际上最大的生物信息中心。**NCBI 拥有上百个数据库和软件工具,包括著名的生物医学文献摘要数据库 PubMed、参考序列数据库 RefSeq等。1989 年,NCBI 接管了核酸序列数据库 GenBank 。
如何将信息中心的序列进行有效搜索?带着这个目的,大卫·李普曼(David Lipman)和同事共同开发了搜索软件 BLAST(Basic Local Alignment Search Tool)。
今天,我们每个从事生物领域的人都在使用,慢慢形成了“国内网速测试用百度,国外网速测试用blast”的习惯。BLAST已经成为序列比对软件代名词。 BLAST经历了20多年的升级,功能、性能一版比一版好。2009年是BLAST发展的关键一年,升级版——BLAST+出现,BLAST+使用了BLAST的核心算法,延 续了BLAST的优势功能:命令行模式功能增强;新增了update_blastdb.pl等程序;将原来的blastall分离成blastn, blastp,blastx等作为独立的程序,便于管理维护
主要分类:
BLAST采用了一种局部序列比对的算法分析两个序列中的相似区域,并计算统计显著性。主要包括四大类:
1. Nucleotide BLAST
blastn: 将待查询的核酸序列及其互补序列放到核酸库中的一种查询,库中存在的每条已知序列都将同所查序列作一对一的核酸序列比对
2. Protein BLAST
blastp: 蛋白序列到蛋白库中进行查询,库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对
3. BLASTX
核酸序列到蛋白库中进行查询:先将待查询的核酸序列按六种可读框架(逐个向前三个碱基和逐个向后三个碱基读码)翻译成蛋白质序列,再去蛋白库对翻译成的每一条序列作一对一的蛋白序列比对
4. TBLASTN
蛋白序列到核酸库查询:先将核酸序列数据库中的核酸序列按六种可读框架翻译成蛋白质序列,然后将待测蛋白序列再与翻译后的核算数据库比对
使用方法
在线比对:
使用最多的就是在线模式了,搜索blast第一个就是
下面以使用最频繁的blastn为例:
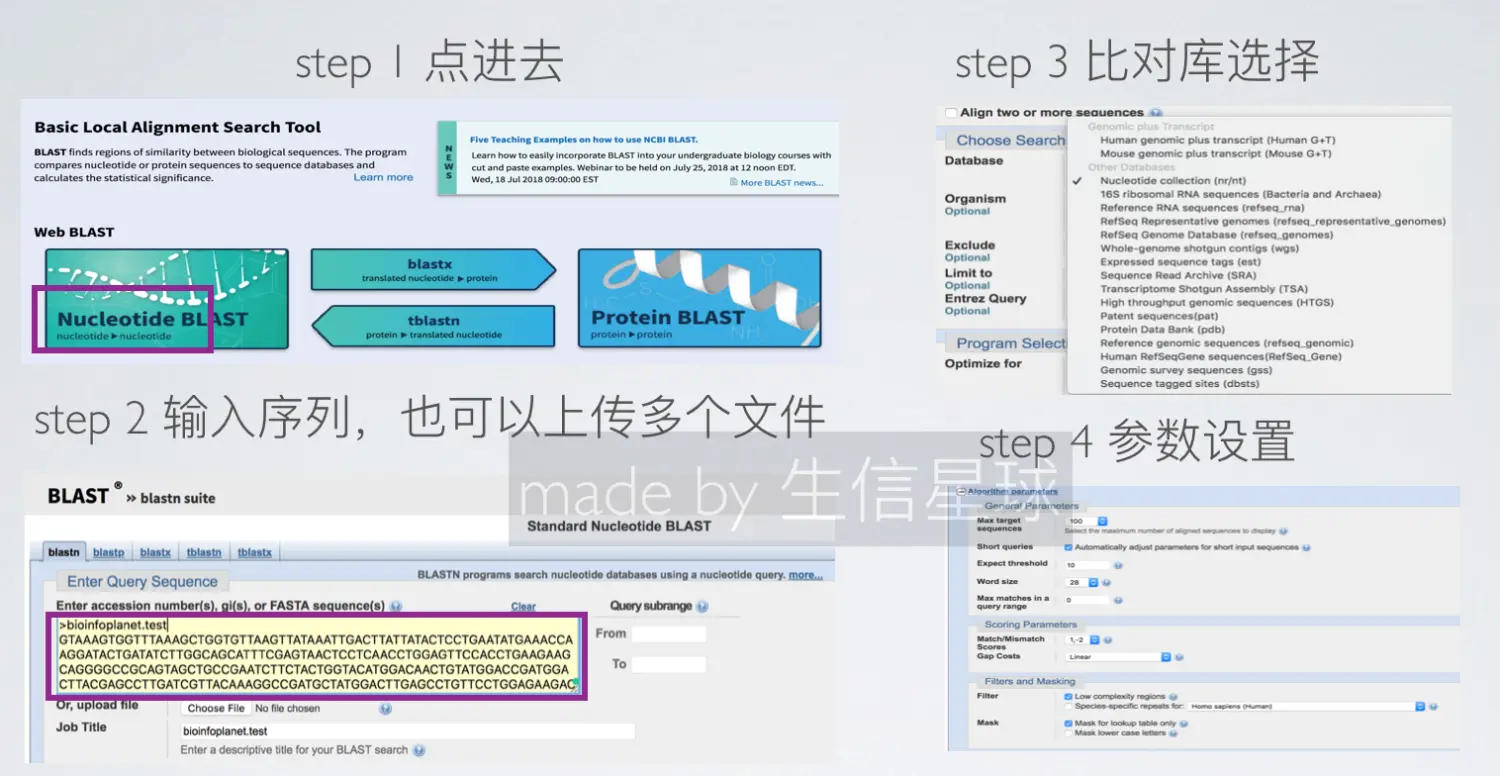
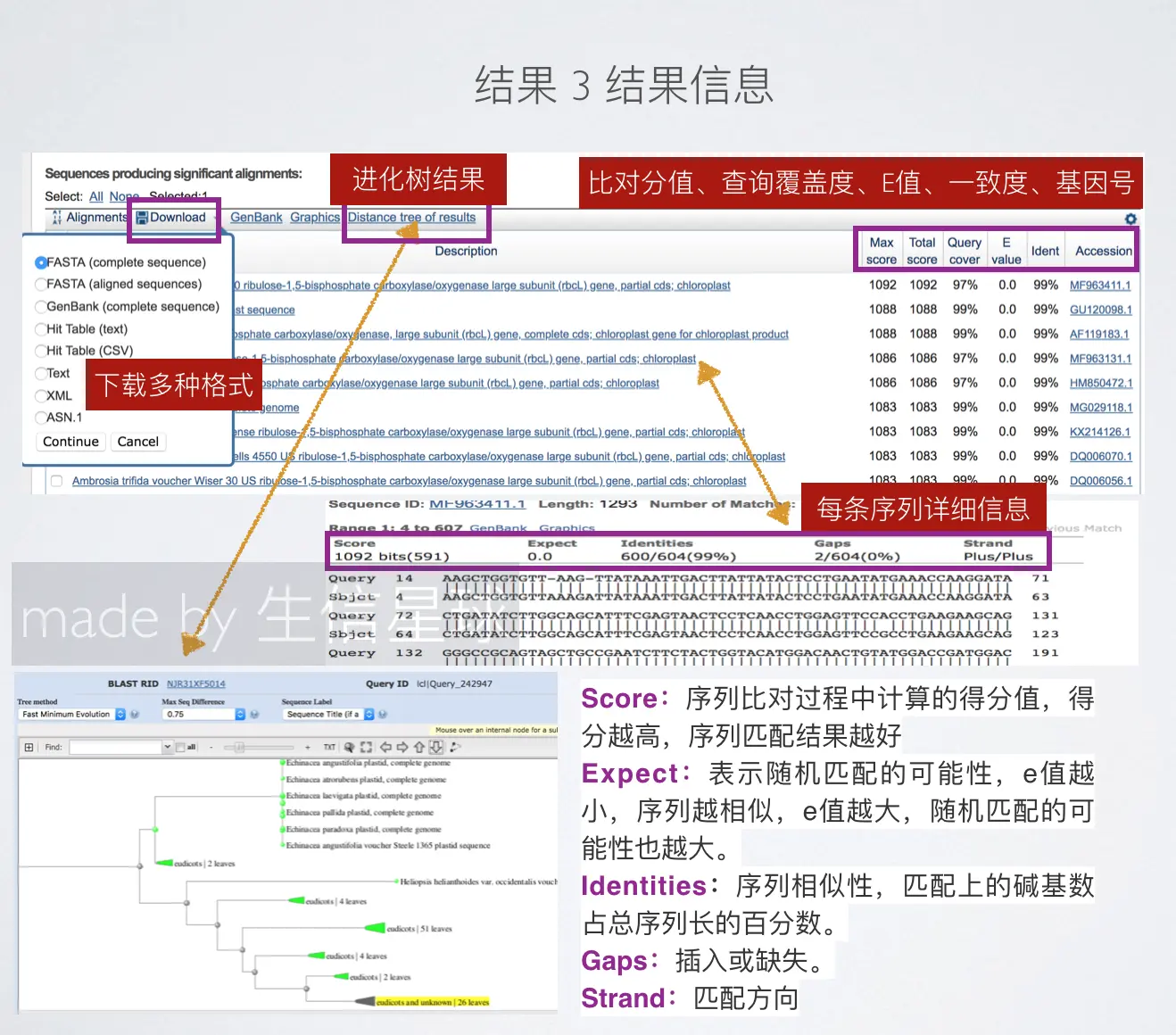
结果显示:
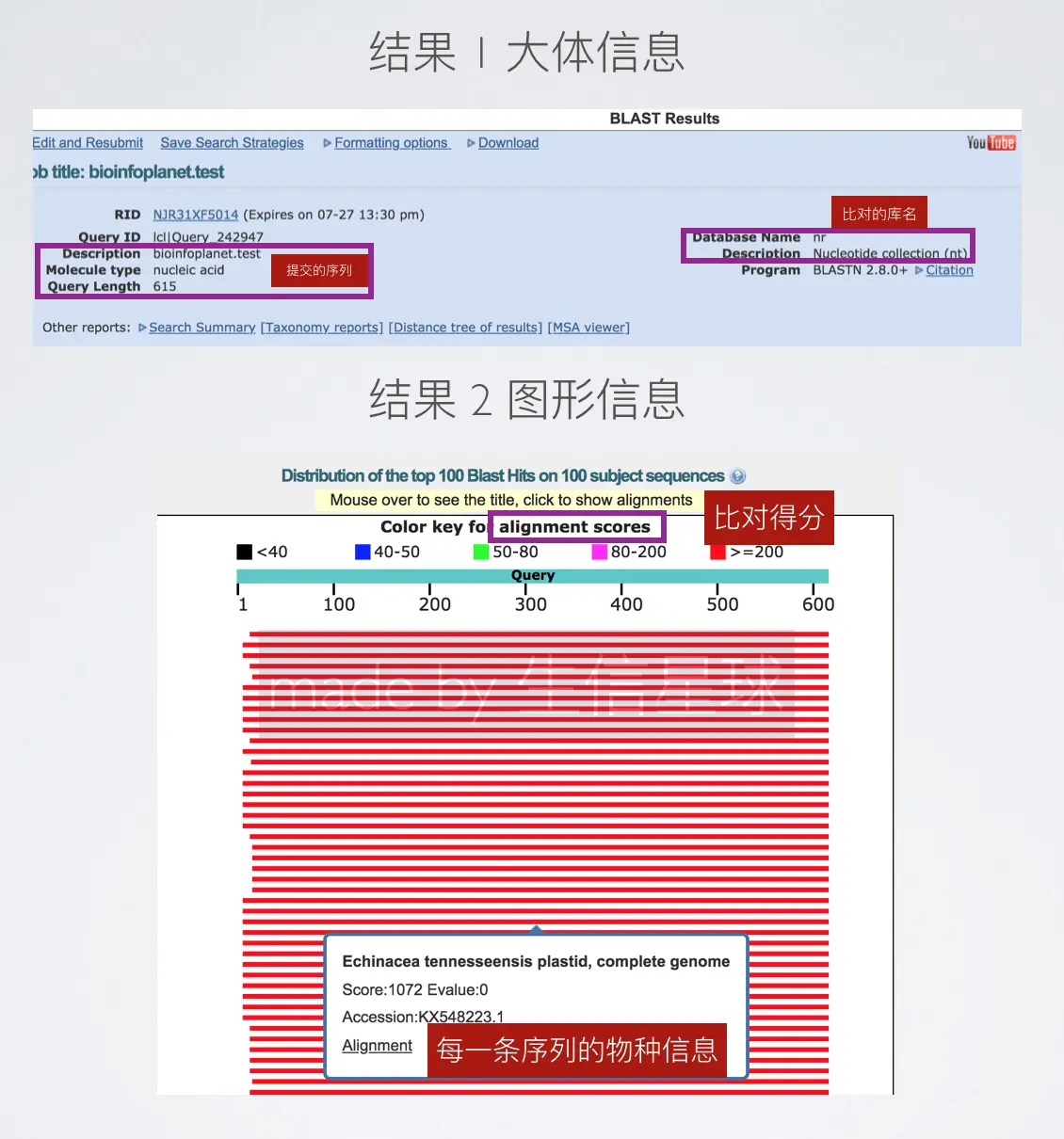
本地比对:
进行序列比对的时候,你有没有遇到过这种问题? NCBI网站太慢,好不容易提交的序列被无情的网站自动刷新给刷没了; 手上序列太多,几百条,手动点击什么时候是个头? 这些机械化的工作,有没有人能帮我做!!!
有的! 让不辞辛苦的服务器帮助你,高效、省时、便利
问个问题,半个小时能干什么?
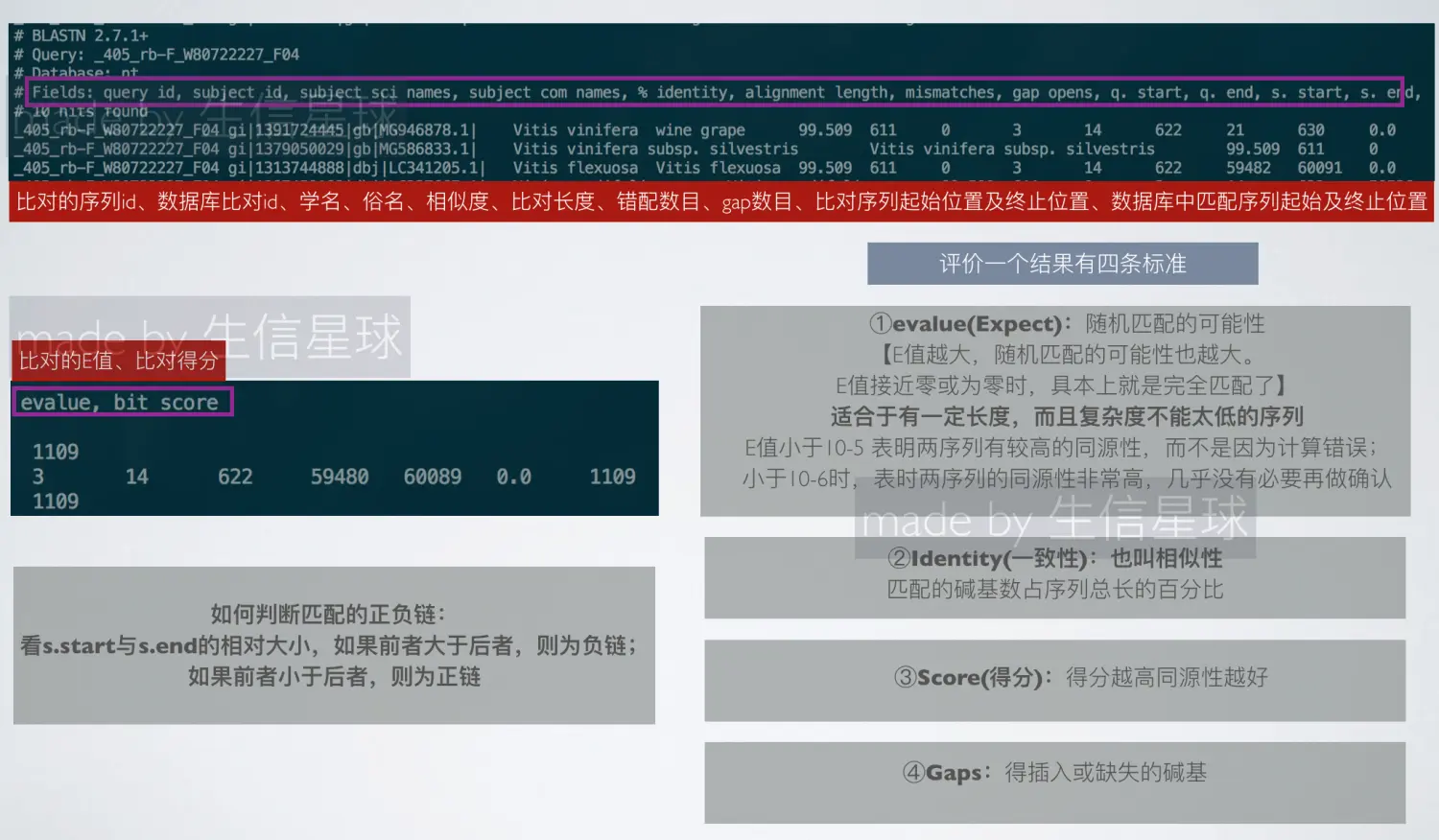
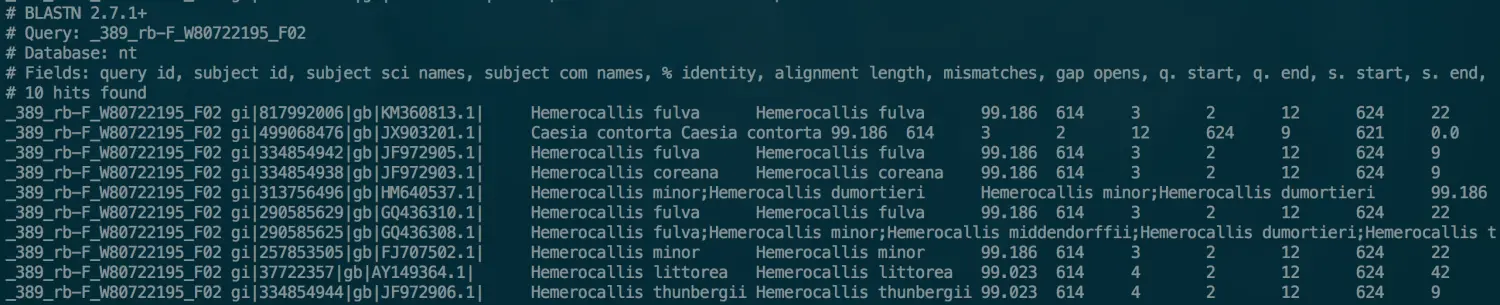
提交十几条序列到NCBI blast?或者做个小小的实验? 又或者~🤨比对500条序列到nt库?中间吃点东西散散步就干完活了【服务器(使用12线程)】 并且结果直接得到像这种~ 基因号、学名、俗名、比对结果信息一目了然
半小时,你买不了吃亏也买不了上当~走过路过,机会难得~【话虽这么说,但豆豆花花不卖,只做分享😍】
如何配置本地blast呢?
第一步,你要有一个服务器或者性能好一点的电脑(台式机、笔记本都可以),推荐linux系统(Ubuntu或CentOS均可)或虚拟机、双系统或git bash,也有windows版本不过豆豆没有用过
第二步,下载blast+软件,下载地址ftp://ftp.ncbi.nlm.nih.gov/blast/executables/LATEST/ncbi-blast-2.7.1+-x64-linux.tar.gz
第三步, 下载nt、nr库
先确保电脑上有300G+空闲空间
两种方式:
- 使用blast+ 自带的
update_blastdb.pl直接运行【优点:简单、日后升级数据库方便;缺点:速度慢,容易卡死】; - wget手动下载,地址
ftp://ftp.ncbi.nih.gov/blast/db【优点:速度快,可断点续传;缺点:日后更新的话需要再次下载】
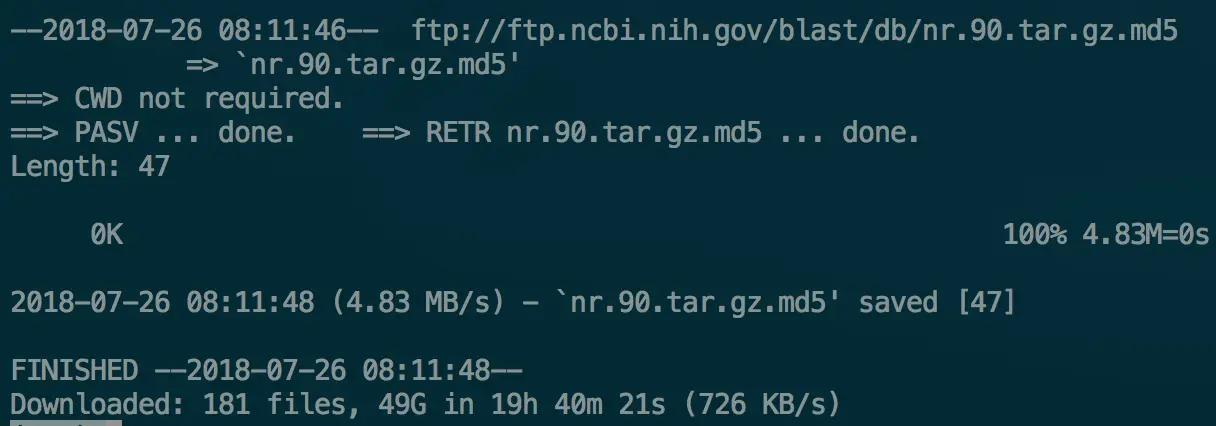
我这里选择的是wget下载方式
这里列出我配置的nr数据库的下载结果,平均速度726KB/s,总大小49G压缩文件,下载时间19h40m21s
第四步, 配置本地.ncbirc文件
# 意思就是指引blast程序去哪里寻找你的数据库
; Start the section for BLAST configuration
[BLAST]
; Specifies the path where BLAST databases are installed
BLASTDB=/home/ncbi/blast/db_nt
; Specifies the data sources to use for automatic resolution
; for sequence identifiers
DATA_LOADERS=blastdb
; Specifies the BLAST database to use resolve protein sequences
BLASTDB_PROT_DATA_LOADER=/home/ncbi/blast/db_nr/nr
; Specifies the BLAST database to use resolve protein sequences
BLASTDB_NUCL_DATA_LOADER=/home/ncbi/blast/db_nt/nt
; Specifies the BLAST database to use resolve protein sequences
BLASTDB_NUCL_DATA_LOADER=/home/ncbi/blast/mydb
BATCH_SIZE=10G
; Windowmasker settings
[WINDOW_MASKER]
WINDOW_MASKER_PATH=//home/ncbi/blast
; end of file
第五步: 转换一下数据格式
拿来数据不要急着分析跑程序,先了解你的数据,看他们怎么命名,思考如何进行修改,前期工作都要把精力放在这里,跑流程是水到渠成的事
一般测序公司返回的数据都是.seq 格式,豆豆处理过一些数据发现,他们每个公司的格式都不统一,命名无规则,十分混乱,常见的有这几种:

转换格式需要一些linux基础知识,会涉及到xargs 、grep、rename、awk、sed命令,简单掌握这几个就可以让数据变成你想要的格式,包括批量替换500个序列的后缀、批量处理文件名中的特殊字符、批量将单纯的序列变为fasta文件(并且能将每个文件名对应进fasta的第一行gene id)等
也可以参考https://mp.weixin.qq.com/s?src=11×tamp=1532587976&ver=1021&signature=ZR-k3-2FShGpW1D8ZoBeWWv-8nGwpxC6kp0jGtp-Wnff4qp0T6*FZZ7KuYwZVpbXppKe-IHjYo79GoeDfR2QbSPCmV615yfs-ClIsgnVOCoMbsUMqU2QB71zhKeizz6p&new=1
全部转换为fasta后,用 cat *.fasta > all_std.fasta ,下面就对all.fasta进行操作
第六步: 运行blast
blast+提供了5个程序
| blastn | 就是进行完全匹配的传统blast |
|---|---|
| blastn-short | 查询短于50个碱基的 |
| megablast | 查询十分相似序列(如物种内或者有关物种) |
| dc-megeblast | 查询关系较远的序列(如物种间) |
| rmblastn | 兼容RepeatMasker |
一个范例程序:
blastn -query all_std.fasta -db nt -outfmt 11 -out "all_std_fm11.blastn@nt.asn" -evalue 1e-5 -perc_identity 99 -num_alignments 10 -num_threads 10
一些参数的设置:
-query: 要比对的序列
-db:比对的数据库
-outfmt:输出格式(共11种)
# 建议先使用-outfmt 11格式,之后可以任意转换成1-10格式【具体格式信息命令行输入blastn -h outfmt】
# 默认是格式0,比较常用的是5、6、7,其中7是带有注释的表格形式
-out: 输出文件名(可以自定义)
-evalue:输出结果的e-value值,一般1e-5
-perc_identity:比对的最低相似度
-num_alignments:输出比对上的数目(默认200),不过一般几百条序列,每一条比对结果有200个,那也看不完,所以这里可以设置前10条
-num_threads:线程数【linux下可以通过一条命令查看电脑或者服务器的线程数】
# cat /etc/proc |grep "process" | wc -l
如果设置了输出格式11,那么后来怎么转换呢?
blast_formatter -archive "all_std_fm11.blastn@nt.asn" -outfmt "7" > "new_all_std_fm7.blastn@nt.asn"
第七步: 查看结果
本地blast应该是比在线版更清楚直观的表现形式了