015-单细胞测序的知识
刘小泽写于18.7.20
介绍
Bulk RNA-seq:
测量一个大的细胞群体中每一个基因的平均表达水平,对比较转录组学(例如比较 不同物种的相同组织) 是有帮助的,但对于研究异质性系统(例如,复杂的组织如脑)还是不够的,对于基因表达的本质研究不够深入
scRNA-seq:
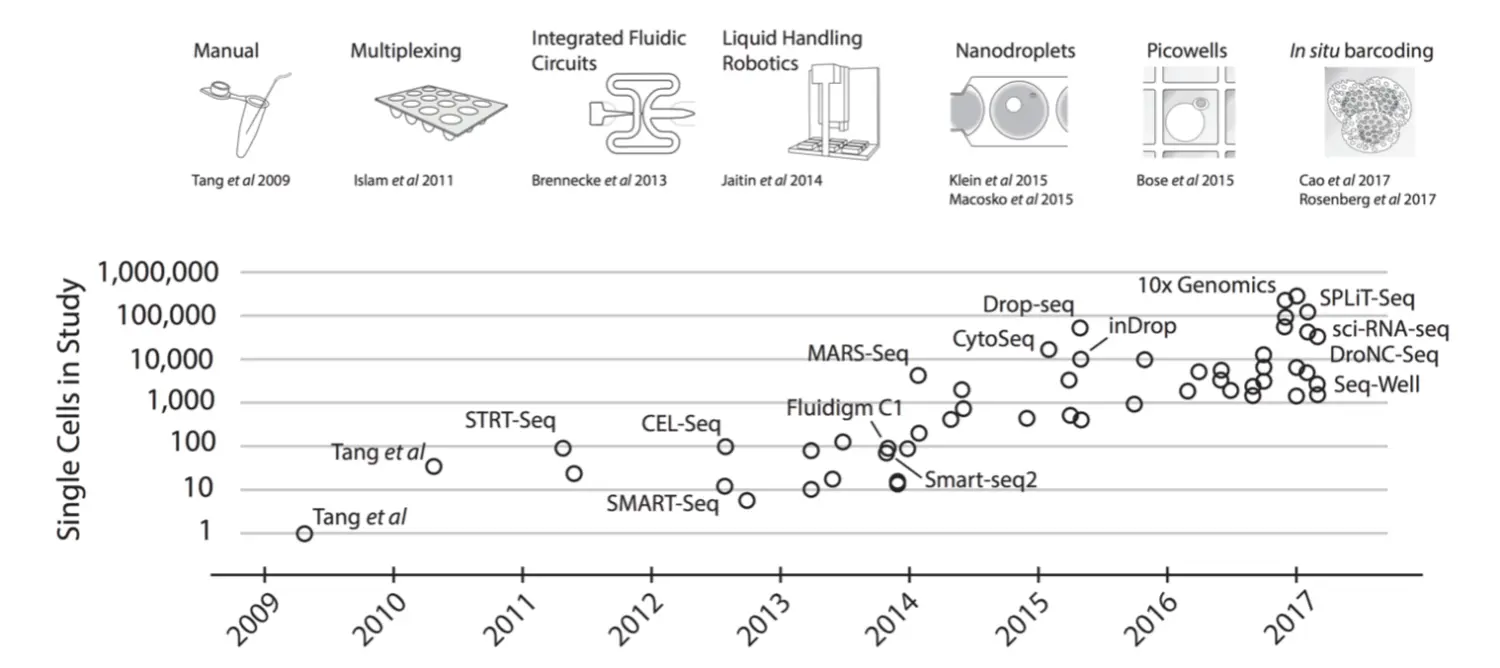
2009年由Tang发表,但直到14年才降低测序费用,逐渐进入大家的视野。它测定的是细胞种群中每个基因的表达量分布,对于研究特定细胞转录组的变化是重要的。测定的细胞量也由最初的10^2^ 涨到了10^6^ 并且还在递增,向着高通量的方向发展。现在有许多处理单细胞测序的流程,比如13年的SAMRT-seq2,12年的CELL-seq,15年的Drop-seq。有一些做单细胞的平台,包括Fluidigm C1、Wafergen ICELL8、10X Genomics Chromium。
单细胞测序的流程:
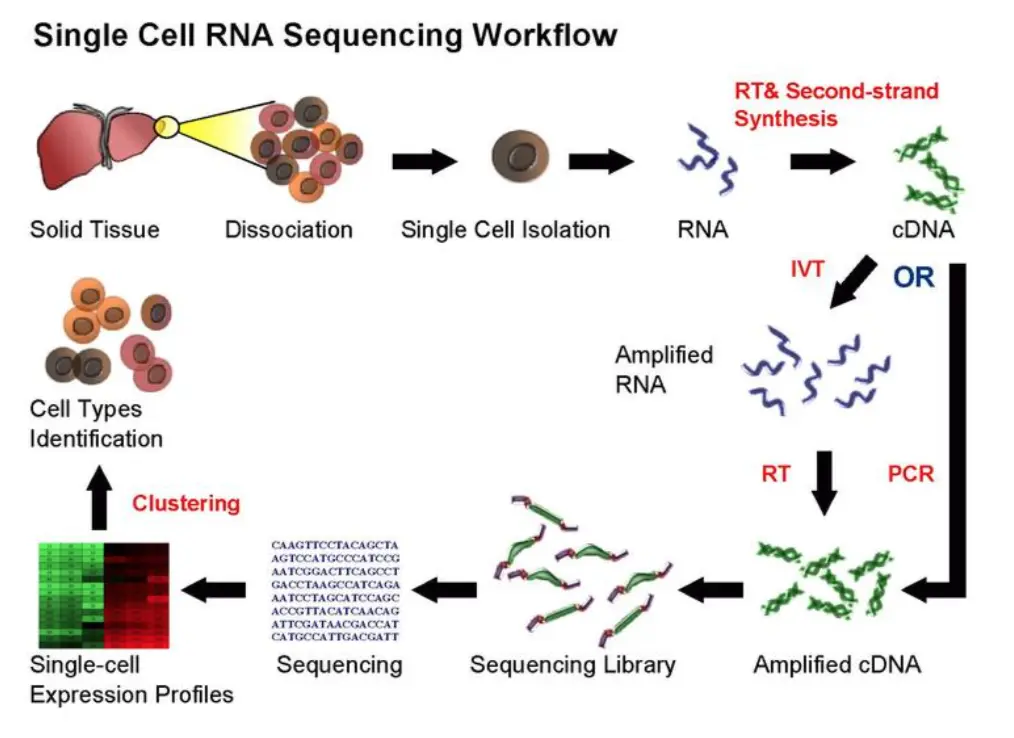
看着和普通的mRNA转录组差不多,取材要求更高了
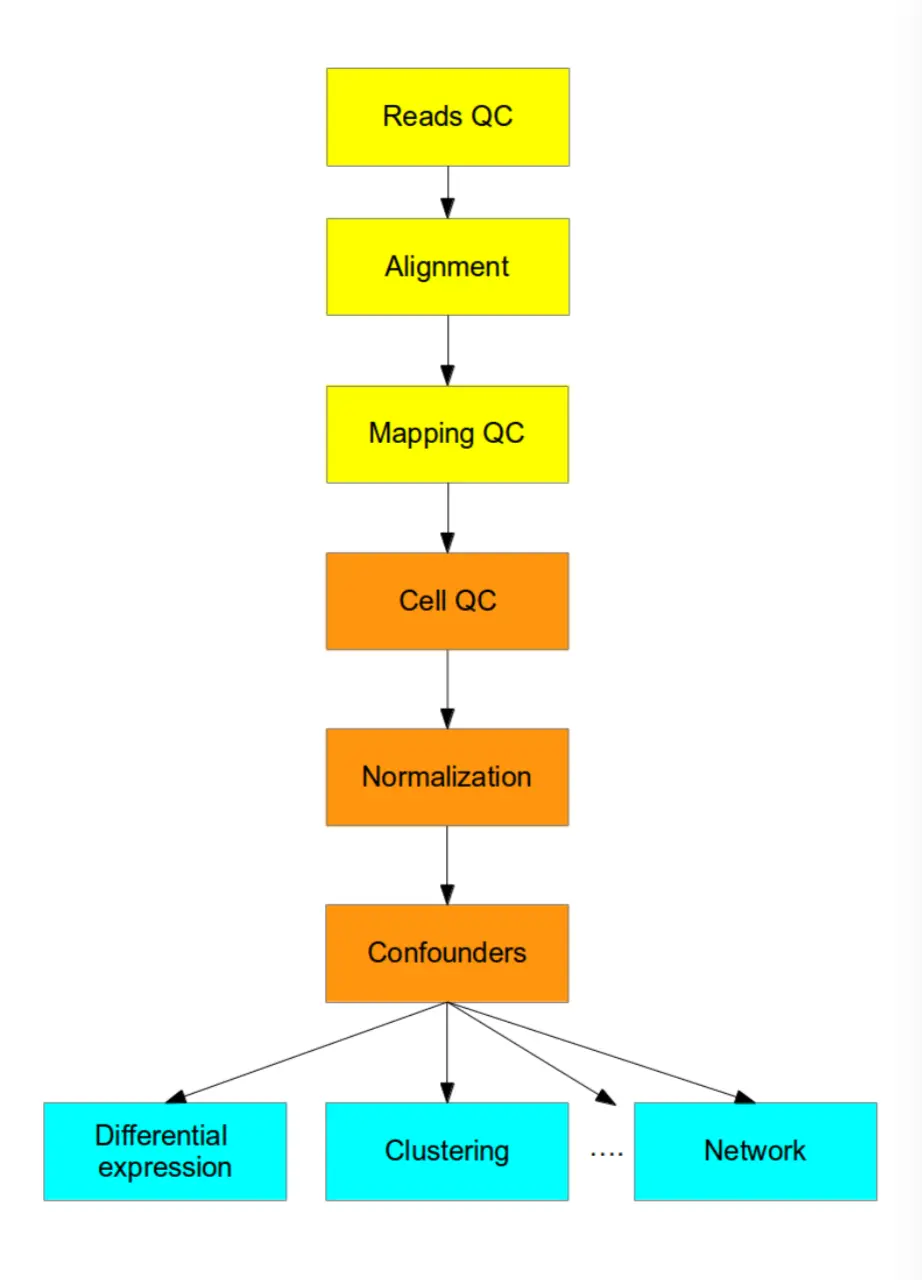
黄色部分对于高通量数据的处理都是差不多的流程; 橙色的部分需要整合多个转录组分析流程以及显著性分析,来解决单细胞测序的技术误差; 最后是下游表达量、通路、互作网络等生物类别的分析,需要使用针对单细胞研发的方法
可以借助许多工具:
- Falco [云端处理流程]
- SCONE(Single-Cell Overview of Normalized Expression)—一个质控和标准化的包
- Seurat– QC以及后续分析探索数据
- ASAP(Automated Single-cell Analysis Pipeline) —交互式网页分析
面临挑战:
单细胞转录组与群体转录组的最大不同之一就是:测序文库是建立在单独一个细胞上的,并非一群细胞。单细胞目前面临的挑战主要是:扩增量目前最多是一百万个细胞;有可能一个基因在一个细胞中能检测到中等表达量,但是在另一个细胞中却检测不到,这种现象叫做"gene dropouts”。下一步需要增加转录本捕获效率,减少扩增误差
分析方法:
近几年,发展起来的方法很多,大体上(并不全面)包括:
- CEL-seq (Hashimshony, 2012)
- CEL-seq2(Hashimshony, 2016)
- Drop-seq(Macosko, 2015)
- InDrop-seq (Klein, 2015)
- MARS-seq(Jaitin, 2014)
- SCRB-seq(Soumillon, 2014)
- Seq-well (Gierahn, 2017)
- Smart-seq (Picelli, 2014)
- Smart-seq2 (Picelli, 2014)
- SMARTer
- STRT-seq (Islam, 2013)
方法主要分为两大部分:定量与捕获。
定量包括两种类型:全长以及基于标签(tag)。前者对每个转录本都试图获得一致的read覆盖度,后者只捕获5‘或者3’端的RNA。定量方法的选择也影响了后续分析的方法选择。理论上,全长的方法应该得到转录本的平均覆盖度,但是实际上,覆盖度经常是有偏差的。基于标签的方法能够利用特异性分子标记(Unique Molecular Identifiers, UMIs)提高定量准确度,但是呢,这种限制了转录组一端的方法有降低了转录本的可拼接性,让以后的isoform识别变得困难。
捕获的技术决定了细胞如何被筛选、获取怎样的测序外的补充信息、数据产量,三种最常用的方法是基于微孔microwell-、微液流microfluidic-、微滴droplet-
基于微孔的捕获技术可以使用细胞移液器或激光捕获分离细胞,并将他们放置于微液流孔中。这样的一个优势就是:可以与荧光触发细胞分离技术(FACS)结合,实现基于表面标记的细胞选择。对于想要分离特定细胞群的研究很有效;另外一个优势就是,可以对细胞进行拍照,帮助确定孔中是否存在损伤的或者重复的细胞。当然,他也有缺点,通量低,对每个细胞进行操作需要很大的工作量。
基于微液流的捕获技术 例如Fluidigm’s C1

相对微孔,它的通量更高。但是他的弱点就是:只有10%的细胞能被捕获到,加入处理的细胞类型比较稀少,那么能被芯片捕获的就更少了,数据产出是不够的。另外芯片相对较贵,但是试剂的价格再降低,日后可能总的价格也会下降。
微滴技术 是将单个细胞包裹在µl级别的液滴中,液滴被搭载到建库所用的酶上,每个微滴包含一个独特的条码(barcode),由那个被包装好的细胞产生的所有reads都被贴上了该条码,也是为了之后对于不同细胞reads的分辨。一般微滴技术的通量最大,查资料得知一秒能包装7万个以上的液滴,并且建库费用合适–0.05美元/细胞。但是高额测序费用又成了他的短板,鉴定出的一般只有一千多个差异转录本,覆盖度还是比较低的
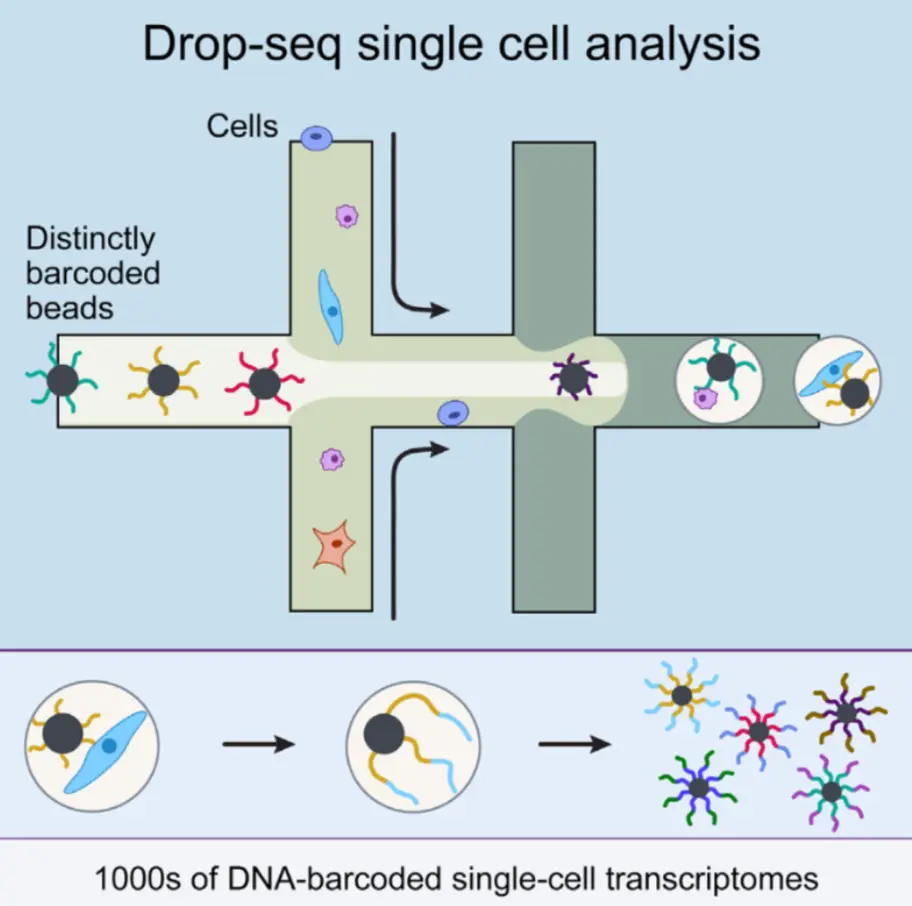
如何根据实验选择测序平台?
**关于捕获,**比如:如果想要分析组织中的成分,那么微滴技术可以提供数量可观的细胞; 如果研究的细胞种群比较稀少,并且有已知的标记,那么最好用FACS,测少量细胞即可; 关于定量,比如:如果想要研究不同的转录本,由于标签是有限的,不可能全部标记完这些转录本,那么全长的转录本定量就更合适; UMIs只能用于使用标签的方法,在基因水平做定量更有优势。
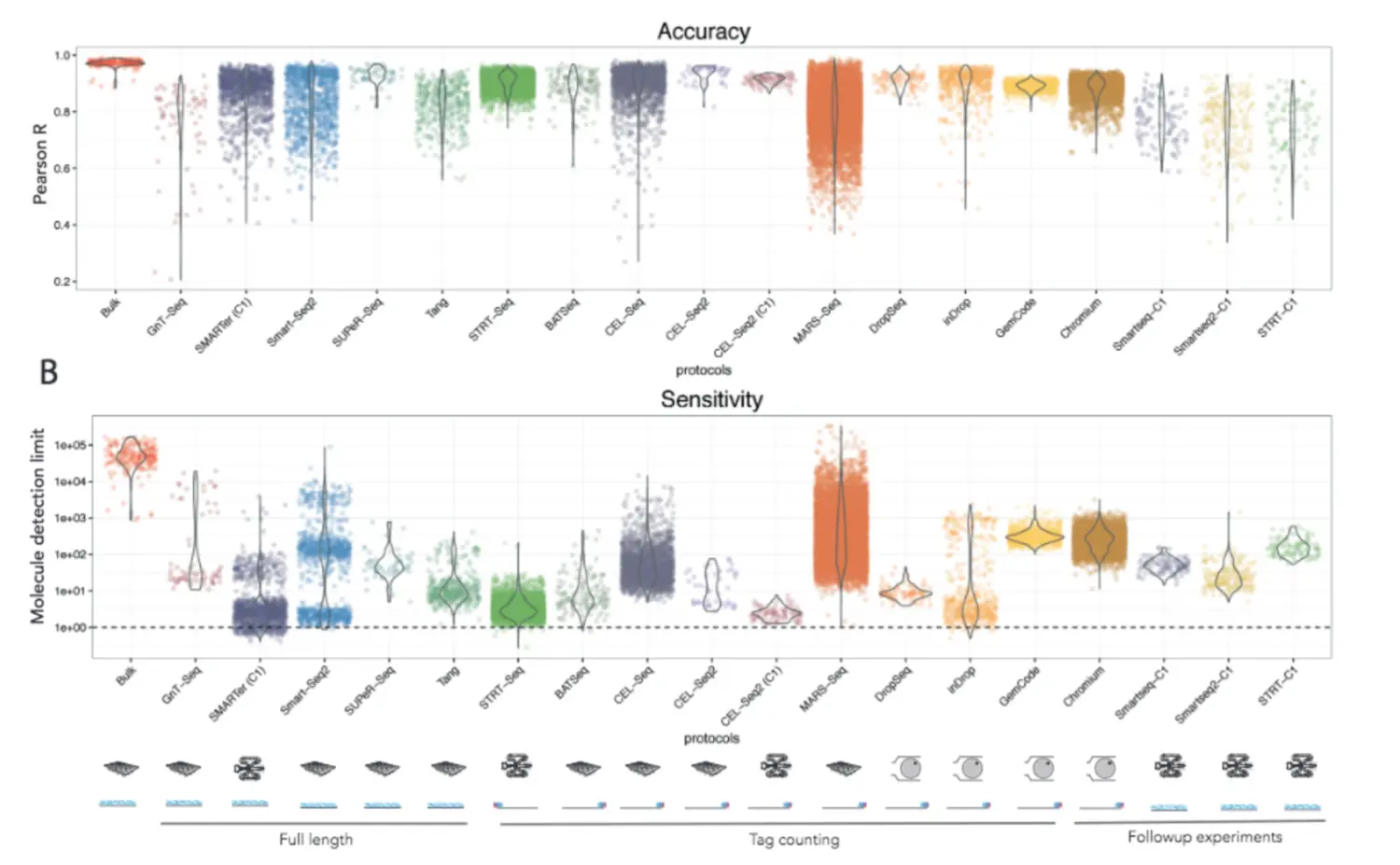
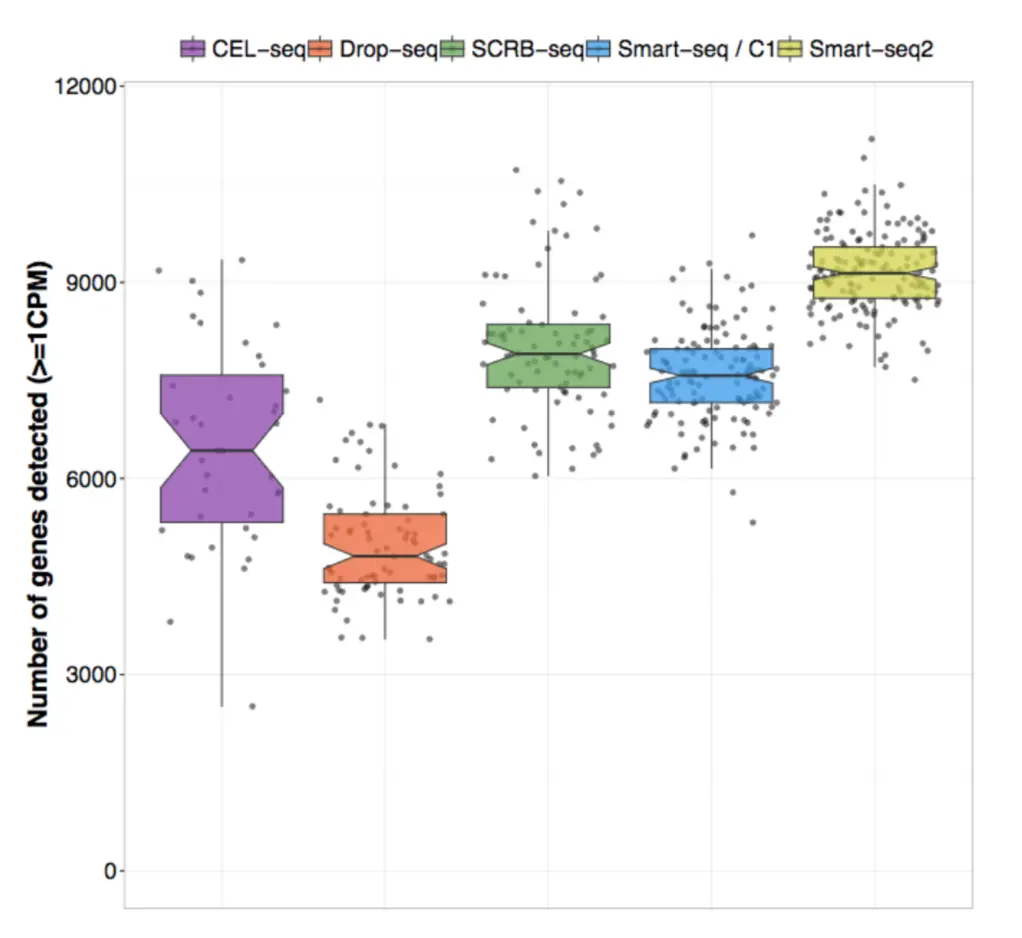
2017年Ziegenhain所在的Enard团队和Svensson所在的Teichmann团队都比较了不同的方法组合:
Ziegenhain使用同样的小鼠胚胎干细胞(mouse embryonic stem cells,mESCs)分析了5种不同的方法,控制细胞数量、测序深度一致,比较了方法的检测灵敏度、噪声水平以及各个方法的费用。结果显示了检测到基因的数目(给定一个检测阈值),结果显示drop-seq检测到的基因数目比Smart-seq2低了两倍。因此,方法的选择对于实验结果很重要!
Svensson使用的是已知浓度的合成的转录本(加入了spike-in)来检测不同方法的准确度和敏感度