129-龙星第二天-alignment
刘小泽写于19.7.30 来北大的龙星计划做助教,接触了一些之前没有了解过的一些知识,简单做下记录,我个人其实是没有这方面需求的 因为课程都是配置在华为云上的,所以使用的数据也都是配置好的,感兴趣的小伙伴其实也不需要自己跑流程(因为用的也都是测试数据),看看怎么使用参数即可
激活conda
如果看到这里的你有linux基础,那么就可以自己配置conda,自主下载安装软件与环境了
如果发现自己账号下面shared这个目录没有内容(出现No such file or directory 可以联系华为云)
# 教程已经将conda封装好,直接激活使用
. /shared/miniconda3/etc/profile.d/conda.sh
conda activate base
进入测试目录
mkdir -p ~/project/alignment/short-reads
cd ~/project/alignment/short-reads
# 然后软链接数据到当前目录(data、ref、vcf)
ln -s /shared/data/NA12878_test_data/short-reads data
ln -s /shared/data/ref_hg37_chr1/ref ref
ln -s /shared/data/ref_hg37_chr1/vcf vcf
以下全部操作都在
~/project/alignment/short-reads目录下进行
质控
看下数据质量
mkdir -p fastqc_output
fastqc -t 2 -o fastqc_output -f fastq data/sr.chr1.2mb_1.fq data/sr.chr1.2mb_2.fq
结果的Html文件用 filezilla导出(选择自己合适的版本进行安装=》 如何使用请看小洁的解答)
(如果不联网的话,可能图片看不了)

对于结果的解读:
短序列比对
测试 reads are from a 2MB region in chr1
看下数据量
wc -l data/sr.chr1.2mb_1.fq data/sr.chr1.2mb_2.fq
# 2432672 data/sr.chr1.2mb_1.fq
# 2432672 data/sr.chr1.2mb_2.fq
# 4865344 total
# fq文件4行为1个read 因此有1.2 million paired-end reads
方法一 minimap2
这款软件也是BWA的作者李恒开发的,它其实更适合于比对PacBio或Nanopore的DNA/mRNA数据
关于程序怎么用,直接命令行输入minimap2
minimap2 -ax sr ref/hg37d5.chr1.fa data/sr.chr1.2mb_1.fq data/sr.chr1.2mb_2.fq | samtools sort | samtools view -bS - > chr1.2mb.mp2.bam
# -ax sr 参数含义: "short genomic paired-end reads"
# 大约1分钟
# 构建索引
samtools index chr1.2mb.mp2.bam
方法二 bwa-mem
bwa mem ref/hg37d5.chr1.fa data/sr.chr1.2mb_1.fq data/sr.chr1.2mb_2.fq | samtools sort | samtools view -bS - > chr1.2mb.bwa.bam
# 大约4min
# 可以加个参数(-t)加速
bwa mem -t 2 ref/hg37d5.chr1.fa data/sr.chr1.2mb_1.fq data/sr.chr1.2mb_2.fq | samtools sort | samtools view -bS - > chr1.2mb.bwa.bam
# 大约2m3.722s
# 索引
samtools index chr1.2mb.bwa.bam
运行命令的同时可以新开一个窗口,登录进去输入top看看运行进程,看到用两个CPU的时候CPU使用量是200%左右
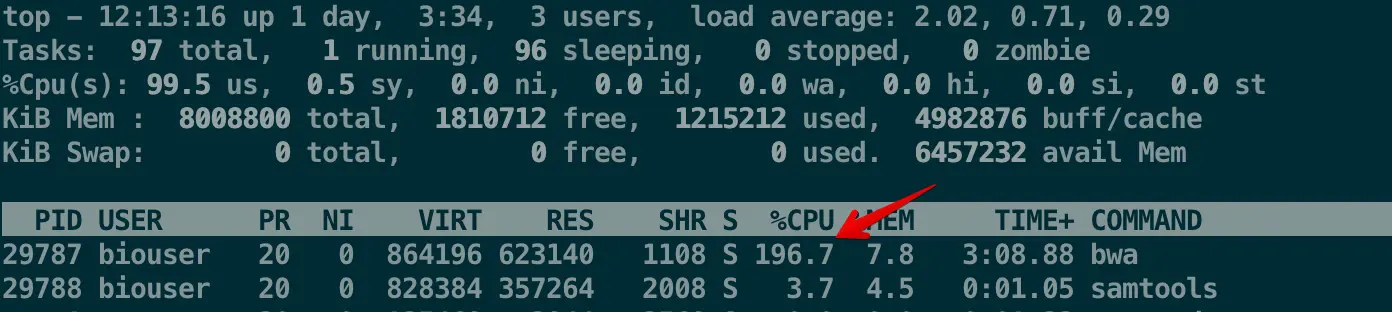
查看bam文件
SAM/BAM文件的格式:https://samtools.github.io/hts-specs/SAMv1.pdf
简单查看
samtools view chr1.2mb.bwa.bam | less -SN # 按q键退出
# 或者直接打印前10行
samtools view chr1.2mb.bwa.bam | head
使用samtools工具
# 格式: samtools tview [options] <aln.bam> [ref.fasta]
# 1.先定义位置,再看
samtools tview -p 1:156000000 chr1.2mb.bwa.bam ref/hg37d5.chr1.fa
# -p chr:pos go directly to this position
# 2.先看,再定义位置
samtools tview chr1.2mb.bwa.bam ref/hg37d5.chr1.fa
# 然后输入g,就会出现一个对话框,输入1:156000211跳转(如下图)
# 退出这个界面,按q

这里就有人问了?为什么我看到的是类似这种
,而且我的还有其他字符,例如既有
,,又有.,又有大写字母,又有小写字母,应该怎么理解,其实简单的搜索就可以解决这个问题:我们只需要搜索samtools tview symbols类似的字符串,就能找到答案 例如在: https://www.biostars.org/p/151739/就有解释:


找变异
主要利用bcftools这个工具,详见:https://samtools.github.io/bcftools/bcftools.html#mpileup

1 对minimap2产出的bam操作
bcftools mpileup -r 1:155000000-157000000 -f ref/hg37d5.chr1.fa chr1.2mb.mp2.bam | bcftools call -mv -Ob -o mp2.bcftools.call.bcf
# 这一步比较慢,大约需要4m25.991s
关于bcftools mpileup
- -r: regions
- -f: fasta-ref FILE
关于bcftools call
-m: multiallelic-caller
-v: variants-only
-O: output-type b|u|z|v (详见:https://samtools.github.io/bcftools/bcftools.html#common_options)
compressed BCF (b); uncompressed BCF (u); compressed VCF (z); uncompressed VCF (v) 那么b和u有什么区别呢?可以看到上面那个文件是用
b参数,下面那个使用u参数得到的,大小差别很大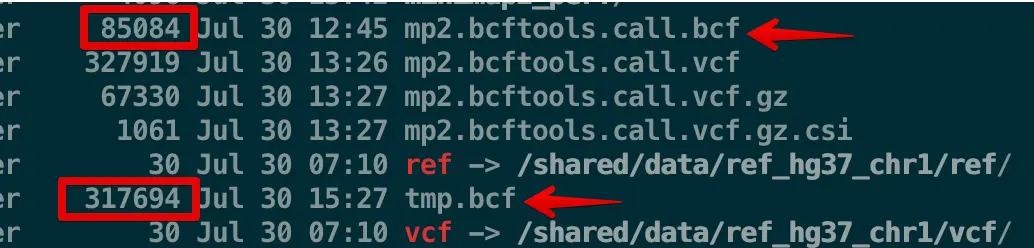
-o:output FILE
然后查看结果
# 直接看
bcftools view mp2.bcftools.call.bcf | less -SN
# 使用过滤条件(如:只看质量值大于20的变异)
bcftools view -i '%QUAL>=20' mp2.bcftools.call.bcf | less -SN
# -i: include EXPRESSION
像这种低质量的就会被过滤掉

因为bcf文件是二进制,有时需要进行转换成文本文件vcf
bcftools view mp2.bcftools.call.bcf > mp2.bcftools.call.vcf
2 对bwa mem产出的bam操作
bcftools mpileup -r 1:155000000-157000000 -f ref/hg37d5.chr1.fa chr1.2mb.bwa.bam | bcftools call -mv -Ob -o bwa.bcftools.call.bcf
bcftools view bwa.bcftools.call.bcf > bwa.bcftools.call.vcf
假如说出现这种报错:
就需要重新运行
samtools index chr1.2mb.bwa.bam这个命令

对变异结果检验
bcftools view -i '%QUAL>=200' mp2.bcftools.call.bcf > mp2.bcftools.call.vcf
bgzip -f mp2.bcftools.call.vcf -c > mp2.bcftools.call.vcf.gz
bcftools index -f mp2.bcftools.call.vcf.gz
bcftools stats mp2.bcftools.call.vcf.gz | grep "^SN"
# 结果SN:Summary Numbers
SN 0 number of samples: 1
SN 0 number of records: 2060
SN 0 number of no-ALTs: 0
SN 0 number of SNPs: 1765
SN 0 number of MNPs: 0
SN 0 number of indels: 295
SN 0 number of others: 0
SN 0 number of multiallelic sites: 0
SN 0 number of multiallelic SNP sites: 0
# 发现:1765 snps and 295 indels among 2 millions bp region
# 也就是约2000/2M = 1/1000,和理论上人类基因组中有千分之一的突变相符
另外看看transitions/transversions的情况
bcftools stats mp2.bcftools.call.vcf.gz | grep "TSTV"
# TSTV, transitions/transversions:
# TSTV [2]id [3]ts [4]tv [5]ts/tv [6]ts (1st ALT) [7]tv (1st ALT) [8]ts/tv (1st ALT)
TSTV 0 1260 505 2.50 1260 505 2.50
其中ts是transitions,也就是转换;tv是transversions也就是颠换
转换:嘌呤和嘌呤之间的替换,或嘧啶和嘧啶之间的替换。 颠换:嘌呤和嘧啶之间的替换。
理论上,全基因组分析中Transition/Transversion比值应该在2左右,全外显子分析中比值在3左右
现在将我们得到的和标准的vcf进行对比
bcftools isec mp2.bcftools.call.vcf.gz vcf/vcf.chr1.2mb.vcf.gz -p minimap2_perf
# bcftools isec [OPTIONS] A.vcf.gz B.vcf.gz […]
# -p: --prefix DIR
# 结果会在minimap2_perf目录中找到四个文件:0000.vcf 0001.vcf 0002.vcf 0003.vcf README.txt
# 其中:0000.vcf 记录了我们得到的vcf:mp2.bcftools.call.vcf.gz
# 0001.vcf 记录了标准的vcf:vcf/vcf.chr1.2mb.vcf.gz
# 0002.vcf 记录了我们得到的vcf中有多少与标准vcf有交集
# 接下来主要用0002.vcf
bcftools stats minimap2_perf/0002.vcf | grep "^SN"
# 结果如下:
SN 0 number of samples: 1
SN 0 number of records: 1588
SN 0 number of no-ALTs: 0
SN 0 number of SNPs: 1569
SN 0 number of MNPs: 0
SN 0 number of indels: 19
SN 0 number of others: 0
SN 0 number of multiallelic sites: 0
SN 0 number of multiallelic SNP sites: 0
# 可以看到这里的交集中有1588个变异,也就是我们检测的变异中有1588个是标准的,之前总共得到了2060个,那么比例就是1588/2060=0.771。如果只考虑SNP的话,就是1569/1765=0.89
关于
bcftools isec生成的4个文件解释:最后两个都是交集为何文件大小差别这么大 https://www.biostars.org/p/224948/ 简单理解就是:因为即使是同一个突变,但在不同的vcf包含的注释信息是不同的
长序列比对
主要使用
minimap2
minimap2 --MD -ax map-ont ref/hg37d5.chr1.fa data/np.chr1.2mb.fq | samtools sort | samtools view -bS - > chr1.2mb.bam
samtools index chr1.2mb.bam
# 关于x的参数说明,有以下几种:
# 1. map-pb/map-ont: PacBio/Nanopore vs reference mapping
# 2. ava-pb/ava-ont: PacBio/Nanopore read overlap
# 3. asm5/asm10/asm20: asm-to-ref mapping, for ~0.1/1/5% sequence divergence
# 4. splice: long-read spliced alignment
# 5. sr: genomic short-read mapping
# 这里的测试数据是nanopore的,所以选择map-ont(ont就是Oxford Nanopore Technologies缩写)
比对完依然是检查bam文件
samtools view chr1.2mb.bam | less
samtools tview -p 1:156000000 chr1.2mb.bam ref/hg37d5.chr1.fa
然后找变异
这里不和短序列一样使用bcftools,而使用了longshot软件,这是一款2019年放出的软件,https://github.com/pjedge/longshot 官方介绍其实很详细了
#先激活一个环境
conda activate longshot
# 它的用法是:longshot [FLAGS] [OPTIONS] --bam <BAM> --ref <FASTA> --out <VCF>
# 然后全部使用默认参数运行
longshot --bam chr1.2mb.bam --ref ref/hg37d5.chr1.fa --out mp2.longshot.o.vcf
同样看看结果
bcftools view -i '%QUAL>=30' mp2.longshot.o.vcf > mp2.longshot.vcf
bgzip -f mp2.longshot.vcf -c > mp2.longshot.vcf.gz
bcftools index -f mp2.longshot.vcf.gz
bcftools stats mp2.longshot.vcf.gz | grep "TSTV"
# TSTV, transitions/transversions:
# TSTV [2]id [3]ts [4]tv [5]ts/tv [6]ts (1st ALT) [7]tv (1st ALT) [8]ts/tv (1st ALT)
TSTV 0 1616 529 3.05 1616 529 3.05
看到TS比TV多了3倍
bcftools stats mp2.longshot.vcf.gz | grep "^SN"
# 结果得到了2145个snp,没有indel
SN 0 number of samples: 1
SN 0 number of records: 2145
SN 0 number of no-ALTs: 0
SN 0 number of SNPs: 2145
SN 0 number of MNPs: 0
SN 0 number of indels: 0
SN 0 number of others: 0
SN 0 number of multiallelic sites: 0
SN 0 number of multiallelic SNP sites: 0
与金标准对比
bcftools isec mp2.longshot.vcf.gz vcf/vcf.chr1.2mb.vcf.gz -p longshot_perf
bcftools stats longshot_perf/0002.vcf | grep "^SN"
# 我们得到的结果与金标准的交叉结果
SN 0 number of samples: 1
SN 0 number of records: 1189
SN 0 number of no-ALTs: 0
SN 0 number of SNPs: 1189
SN 0 number of MNPs: 0
SN 0 number of indels: 0
SN 0 number of others: 0
SN 0 number of multiallelic sites: 0
SN 0 number of multiallelic SNP sites: 0
原来短读长比对金标准结果总共得到了1588个变异,这里长读长得到了金标准1189个,计算recall ratio= 1189/1588=0.75;然后我们自己计算得到了2145个,金标准有1189个,计算precision rate = 1189/2145=0.55 ,而这个比例在短读长里面是0.7以上的,因此长读长这个方面略显劣势
那么短读长和长读长之间有多少检测一致呢?
bcftools isec ../short-reads/mp2.bcftools.call.vcf.gz mp2.longshot.vcf.gz -p shortlong_comparison
bcftools stats shortlong_comparison/0002.vcf | grep "^SN"
# 结果看到有1294个交集
SN 0 number of samples: 1
SN 0 number of records: 1294
SN 0 number of no-ALTs: 0
SN 0 number of SNPs: 1294
SN 0 number of MNPs: 0
SN 0 number of indels: 0
SN 0 number of others: 0
SN 0 number of multiallelic sites: 0
SN 0 number of multiallelic SNP sites: 0
因此对于长读长reads进行variant calling操作,还需要更复杂的工具来做,比如谷歌的 DeepVariant 、Ruibang Luo开发的 Clairvoyante