126-2019意犹未尽的基因组可视化IGV-2
刘小泽写于19.6.16 上一次写了第一部分(熟悉IGV):https://www.jianshu.com/p/9221ce6c5092 这次从实战开始加深印象
前言
记得之前有人问过,IGV下载的基因组在哪里?
IGV其实在安装过程中是在主目录下新建一个叫igv的文件夹的,其中包含了几个重要的组件:
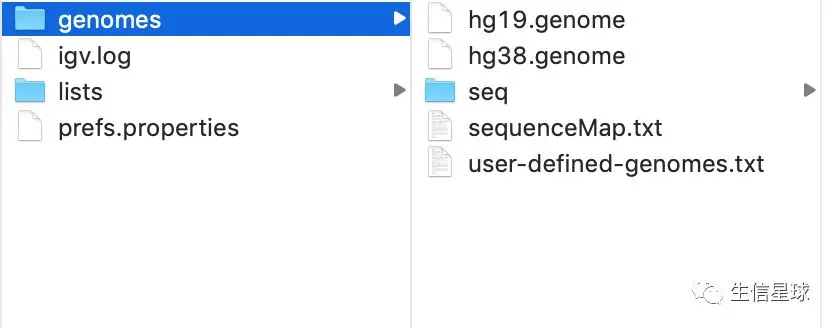
igv.log存储IGV运行的正常和错误日志(这个文件是可以重置的,可以删除或重命名,但需要先退出IGV)prefs.properties存储所有偏好设置(也即是View=>Preferences中的设置),因此在不同的IGV窗口都会打开一样的界面,保持一致性(同样可以删除或删除这个文件,用来重置)genomes目录存储所有下载的基因组文件
另外,可不可以使用自己下载的基因组
是可以的,只需要通过Genomes=>Load Genome form File,导入FASTA文件,但是最好先用samtools 的faidx构建一个.fai索引文件(其实如果IGV找不到的话,它也会自己尝试去构建一个索引,但还是自己先构建好比较把握)
第二部分:实战练习
关于数据
这次将会利用breast cancer cell line HCC1143来可视化比对结果,为了速度我们只选择chr21的一段(19M:20M)
要下载的数据:https://xfer.genome.wustl.edu/gxfer1/project/gms/testdata/bams/hcc1143/HCC1143.normal.21.19M-20M.bam
注意:bam文件是测序数据比对结果的二进制文件,默认是生成sam的,但是sam文件过大,因此转换了一下。bai是bam文件的索引文件,bai必须和bam放在同一个文件夹中,但是导入的时候,只需要导入bam即可
当加载一个bam文件时,一般会创建三个相关的tracks:http://software.broadinstitute.org/software/igv/alignmentdata
- Alignment Track 对每个比对的reads进行可视化
- Coverage Track 对测序深度和覆盖度进行可视化
- Splice Junction Track 对可变剪切位点进行可视化
默认仅显示Alignment Track和Coverage Track,可以在track这里右键设置是否显示这几个track
刚导入是这样
因为这一段是chr21的比对结果,因此我们在导航栏中直接输入:chr21:19,480,041-19,480,386 看看结果
如果不进行任何设置,比对的结果是灰色的:

处理灰色,我们还能注意到有白色的条形,白条表示MAPQ(也就是mapping quality为0,即没有比对上);灰色区域很多都是60,表示比对错误率为10^-(60/10)^ = 10的负六次方 ,精确度为99.9999%
导入后的调节
有很大可能,自己在导入后就是有颜色的了。这是因为IGV会自动加上某些设定,方便用户的使用。如果没有颜色,怎么去调节呢?或者又有哪些新的设定呢?
**颜色:**设置color alignments为read strand(可以用来区分正负链)
**新设定:**在bam这个track使用右键,将Sort alignments设置为start location,将Group alignments设置为pair orientation
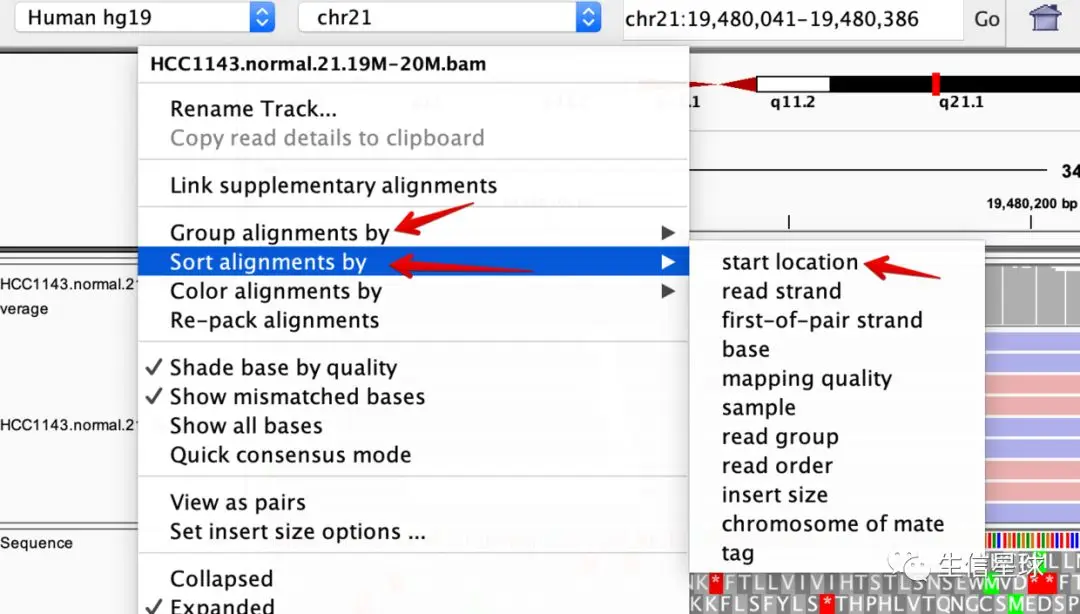
**注意:**比对的结果我们只会看到冰山一角,如果我们继续向下看(最右边是有个下拉条的,拉动它直到底部),就会明白顶栏的峰和谷与比对的reads数量有什么关系

显示read信息: 有三种选项可以设置:悬停显示(Hover)、点击显示和不显示

关于IGV显示的颜色
如果发现这个read中存在与参考序列不同的碱基,会用不同的颜色标注出来(A为绿色,C为蓝色,G是橙色,T是红色)
就像这个位置:chr21:19,480,450-19,480,520 存在G变A的情况
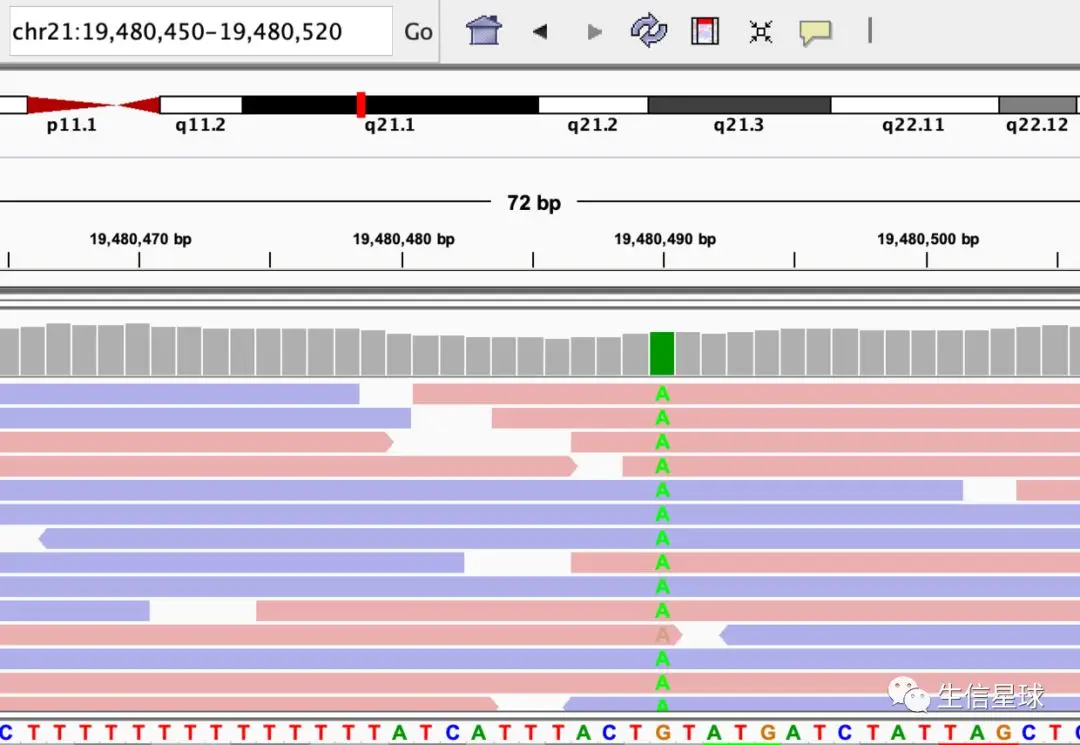
另外,你可能会发现,为什么同样是检测出来的A,颜色还有深浅?
透明度越高,说明检测的碱基质量越低,越不可信

官方文档也给出的答案:http://software.broadinstitute.org/software/igv/book/export/html/37
By default, read bases that match the reference are displayed in gray. Read bases that do not match are color coded, and insertions and deletions within reads relative to the reference are marked. Insertions are indicated by a purple I (
) and deletions are indicated with a black dash (–). In addition, mismatched bases are assigned a transparency value proportional to the read quality known as the phred score. This has the effect of de-emphasizing low quality reads.
然后再来一个练习=》两个相邻的SNPs
第一步:定位到chr21:19,479,237-19,479,814
首先最明显的就是两个杂合子(heterozygous variants),另外结合dnSNP数据库(我们之前利用file=>load from server=>dbsnp1.4.7)可以看到右边👉的杂合子G/T可以对应到数据库(rs982274 );左边的杂合子C/T并没有对应到数据库

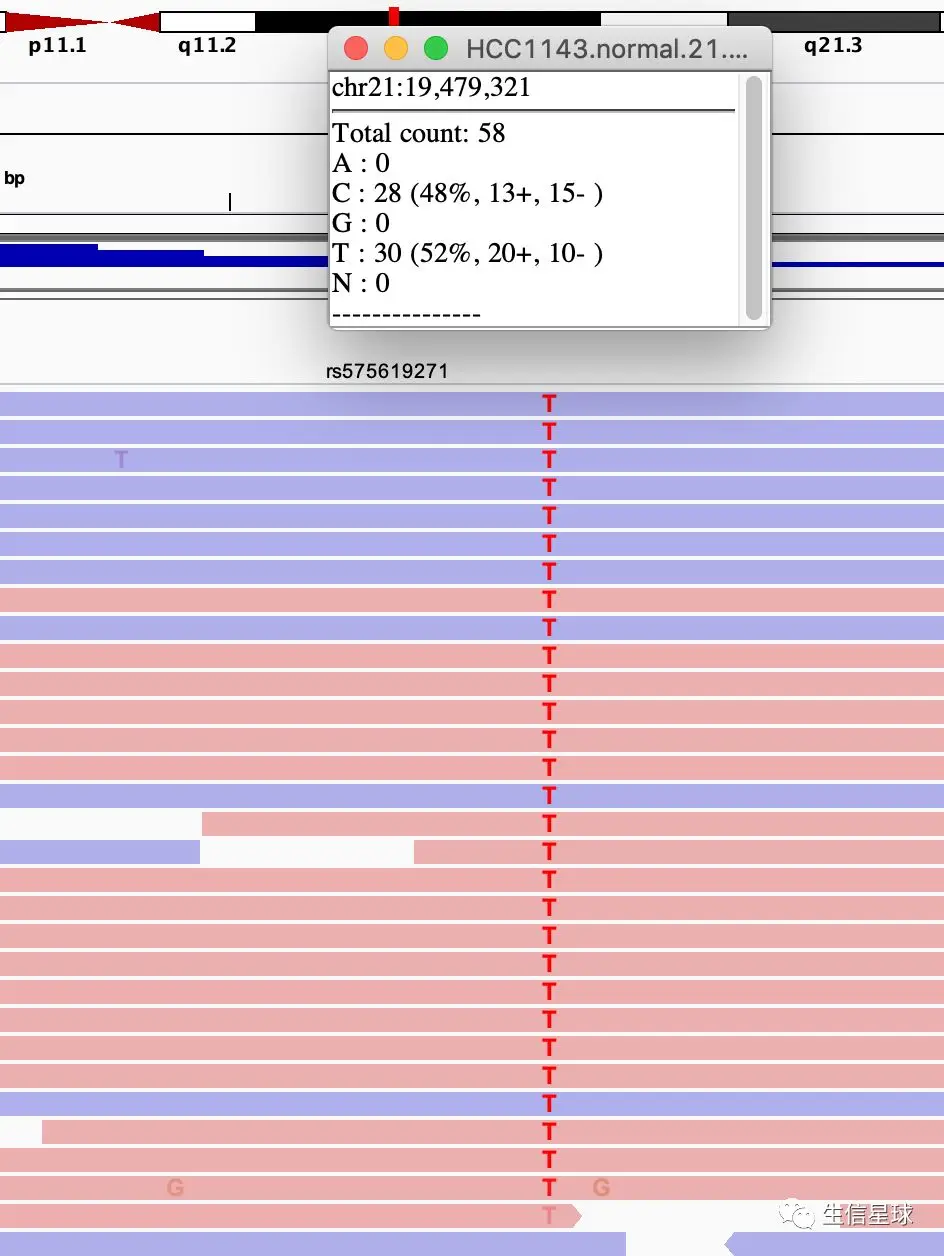
第二步:放大左边的C/T 这个SNV,或者输入这个坐标chr21:19,479,321
然后根据碱基来排序alignment结果,另外利用链区分颜色,得到如下:

另外,从这个图中还能看到:
最后一个检测到T变异read中,碱基质量并不好(因为是透明的)
比对的总体质量不错,并且没有链偏差(strand bias),基因频率和杂合突变结果一致