113-Grep的再次认识
刘小泽写于19.5.8 很久之前写过关于grep的知识, 生信Linux文本处理三剑客 。使用了一段时间后,又有一些更深入的体会
grep -v 进行过滤
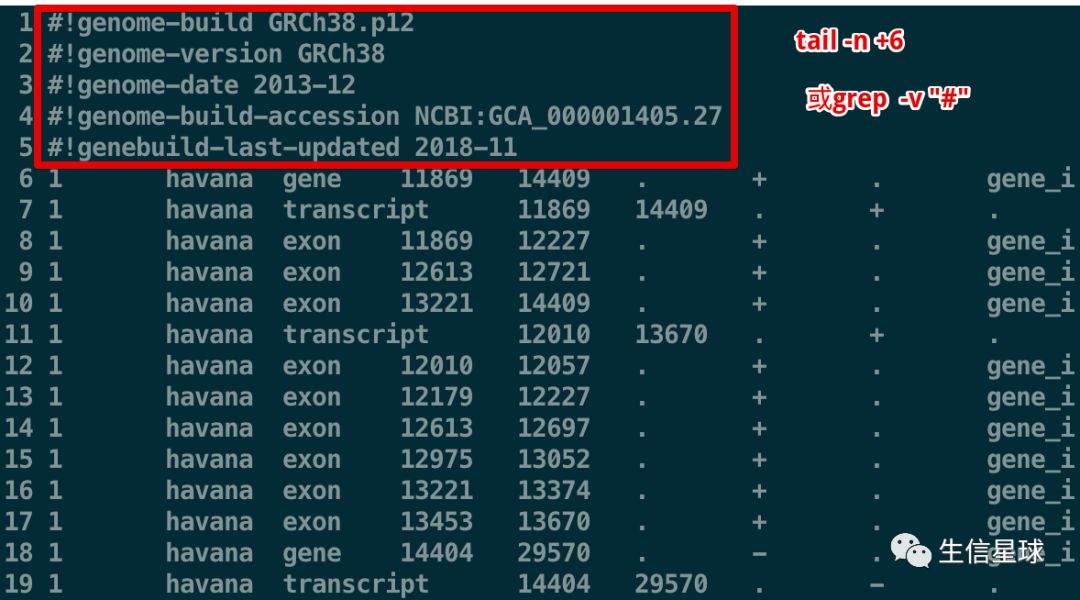
一般拿到gtf文件,不管下面要进行awk还是cut操作,头文件都限制了我们的进步,因此常常使用grep -v 删除头信息,这个比tail表现的更出色,因为它完成了匹配+删除,而tail需要自己去指定删除第几行,比如有5行头文件
使用tail就需要从第6行开始取tail -n +6 Homo_sapiens.GRCh38.96.gtf | head ,而grep直接去掉#行就好了grep -v "#" Homo_sapiens.GRCh38.96.gtf | head
如果现在手头上还没有gtf文件的话,先下载一个
wget ftp://ftp.ensembl.org/pub/current_gtf/homo_sapiens/Homo_sapiens.GRCh38.96.gtf.gz
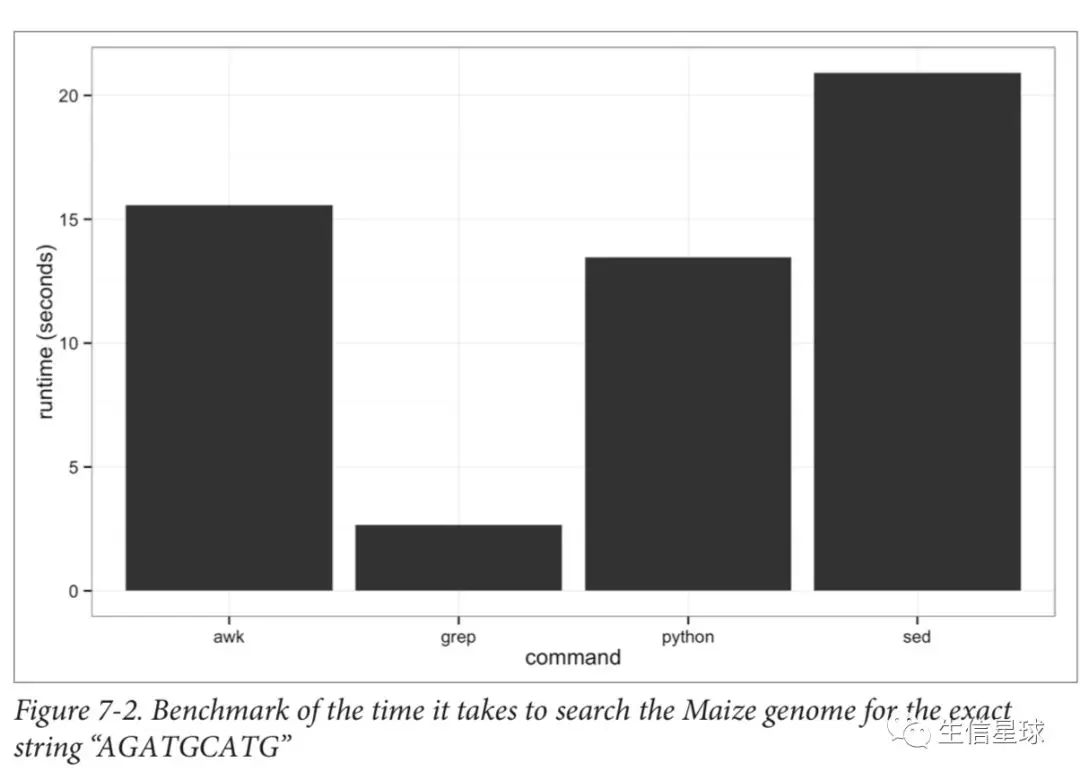
处理速度真的很快
文中就给出了一个测试,利用grep、awk、sed、python脚本去找基因组中某一条序列,但是这样比又有点不公平,因为grep本来就是单线程工作的,如果处理大量的序列搜索,结果就不得而知了(图片来自书籍Bioinformatics Data Skills)
简单搜索
grep一般接受两个参数:匹配格式(包括想要搜索的字符串或者正则表达式)、文件(可以是多个)
下面进行测试(p.s. 因为书中使用的文件是小鼠的1号染色体的基因id文件,并没有给出链接,那么我们就自己构建一个人的hg38的1号染色体的gene id,只是为了测试grep用)
$ awk '{if($1=="1" && $19=="gene_name") {print $10"\t"$20}}' Homo_sapiens.GRCh38.96.gtf | tr -d '";'|uniq >hg38_chr1_gene.txt
# tr -d 的目的是将输出的字符串引号以及分号去掉
$ head hg38_chr1_gene.txt
ENSG00000223972 DDX11L1
ENSG00000227232 WASH7P
ENSG00000278267 MIR6859-1
ENSG00000243485 MIR1302-2HG
ENSG00000284332 MIR1302-2
ENSG00000237613 FAM138A
ENSG00000268020 OR4G4P
ENSG00000240361 OR4G11P
ENSG00000186092 OR4F5
ENSG00000238009 AL627309.1
然后如果要找ZNF692这个基因名
$ grep "ZNF692" hg38_chr1_gene.txt
ENSG00000171163 ZNF692
# 其中的引号可以去掉,但是加上会更安全,因为这明确表示是一个字符串,而不是其他类似的符号
即使部分匹配也会输出结果
$ grep ZNF hg38_chr1_gene.txt| head -n5
ENSG00000125945 ZNF436
ENSG00000249087 ZNF436-AS1
ENSG00000142684 ZNF593
ENSG00000176083 ZNF683
ENSG00000160094 ZNF362
如果要去掉其中的某一个,比如ZNF593
grep ZNF hg38_chr1_gene.txt| grep -v ZNF593
grep -w 精确匹配
遇到上面这种情况要小心,因为-v这里是不完全匹配,很有可能会匹配到类似ZNF5934 、ZNF593A 等情况(这里只是举例,并不代表存在这种真实基因),那么如果我就想去掉ZNF593这一个基因,那么就需要用到-w (即word,按单个词匹配)
为了领会-w的含义,举个例子:
$ cat example.txt
bio
bioinfo
bioinformatics
computational biology
$ grep -v bioinfo example.txt
bio
computational biology
$ grep -v -w bioinfo example.txt
bio
bioinformatics
computational biology
输出上下文
另外,grep有三个非常好用的参数,可以直接将匹配内容的上、下几行输出,它们分别是:
# -B => before,输出匹配内容之前的几行
# -A => after,输出匹配内容之后的几行
# -C => before & after, 输出匹配内容前后几行
$ grep -B1 -w bioinfo example.txt
bio
bioinfo
$ grep -A2 -w bioinfo example.txt
bioinfo
bioinformatics
computational biology
$ grep -C1 -w bioinfo example.txt
bio
bioinfo
bioinformatics
grep与正则
grep是支持BRE(Basic Regular Expression)的,但这里的正则表达没有编程语言(Perl、Python)中的那么强大
例如,现在想看看基因ZNF644与ZNF124的Ensembl ID,可以看出它们有共同的部分ZNF64,这样就可以写最基本的匹配式
$ grep ZNF64[4,8] hg38_chr1_gene.txt
ENSG00000122482 ZNF644
ENSG00000179930 ZNF648
但事实上,我们想匹配的内容一般不会这么简单,比如像匹配ZNF644和ZNF236呢?再用上面的式子去描述就不会那么简单了,这时,更方便的ERE(Extended Regular Expressions)就出来了,使用-E参数调用
$ grep -E "(ZNF644|ZNF124)" hg38_chr1_gene.txt
ENSG00000122482 ZNF644
ENSG00000196418 ZNF124
或者更高级一点的写法是:将grep -E写成egrep,加上忽略大小写-i,加上颜色区分--color
$ egrep -wi --color 'ZNF644|ZNF124' hg38_chr1_gene.txt
grep -c 统计行数
最简单的一个应用就是:别人问你,服务器配置怎样啊,几核的?此时你就应该快速输入下方两个命令的其中一个
# 第一种方法
cat /proc/cpuinfo | grep process | wc -l
# 第二种方法(改进版)
grep -c process /proc/cpuinfo
可以看看有多少以ZNF开头的基因:
$ grep -c "ZNF" hg38_chr1_gene.txt
26
# 这就相当于先grep,然后wc -l统计一下
另外这个-c参数还可以这样用:比如要统计gtf文件中有多少小核RNA(snRNA)
$ grep -c 'gene_biotype "snRNA"' Homo_sapiens.GRCh38.96.gtf
5730
注意如果我们要找的pattern中包含双引号的字符,那么这时就要用单引号表示pattern
grep的输出
grep之所以有很快的匹配速度,是因为它一旦发现了某一行有匹配项,它就不会再看这一行的剩余部分了,然后就直接把这一整行输出,但是有时我们不想将整行都输出,只想输出匹配成功的内容,那么就用-o
$ grep -o "ZNF.*" hg38_chr1_gene.txt | head -n3
ZNF436
ZNF436-AS1
ZNF593
另外GTF的第9列很特殊,是键值对排列,例如gene_id "ENSG00000223972";
,如果只想挑出来这部分,那么可以这样:
$ egrep -o 'gene_id "\w+"' Homo_sapiens.GRCh38.96.gtf | head -n5
gene_id "ENSG00000223972"
gene_id "ENSG00000223972"
gene_id "ENSG00000223972"
gene_id "ENSG00000223972"
gene_id "ENSG00000223972"
发现虽然输出了键值对,但是这结果也太冗余了吧…
这是因为GTF虽然gene id是一个,但是可能存在gene信息、transcript信息、包含的不同的exon信息,于是可以将gene id去重复、排序后输出
$ egrep -o 'gene_id "\w+"' Homo_sapiens.GRCh38.96.gtf | uniq | cut -d " " -f2 | tr -d '"' | sort | uniq >hg38_gene_id.txt
$ head -n5 hg38_gene_id.txt
ENSG00000000003
ENSG00000000005
ENSG00000000419
ENSG00000000457
ENSG00000000460