103-Bioconductor没想象的那么简单-part5
刘小泽写于19.4.11
part1:https://www.jianshu.com/p/b030d05a80a4 part2:https://www.jianshu.com/p/c4fdab9929db part3:https://www.jianshu.com/p/f43d9d07a4af part4:https://www.jianshu.com/p/4560a29d8da2
AnnotationHub
主要存储了一些公共数据和注释信息
library(AnnotationHub)
options()$BioC_mirror ##查看使用bioconductor的默认镜像
# 检查是否存在新版本,先赋一个值,有更新则自动更新
ahub <- AnnotationHub() #目前最新版本是2018-10-24
# 看看hub中都包括什么内容
> names(mcols(ahub))# 看看hub中都包括什么内容
[1] "title" "dataprovider" "species"
[4] "taxonomyid" "genome" "description"
[7] "coordinate_1_based" "maintainer" "rdatadateadded"
[10] "preparerclass" "tags" "rdataclass"
[13] "rdatapath" "sourceurl" "sourcetype"
公共数据提供者dataprovider
unique(ahub$dataprovider) #公共数据提供者
table(ahub$dataprovider)#查看每个提供者都有多少数据
其中包含了多少物种基因组?
> head(unique(ahub$species))
[1] "Homo sapiens" "Vicugna pacos" "Dasypus novemcinctus"
[4] "Otolemur garnettii" "Papio hamadryas" "Papio anubis"
> length(unique(ahub$species)) #包含了多少物种基因组
[1] 2042
hub提供哪些类的信息呢?
> table(ahub$rdataclass) #提供哪些类的信息
AAStringSet BigWigFile
1 10247
biopax ChainFile
9 1113
data.frame data.frame, DNAStringSet, GRanges
40 3
EnsDb GRanges
678 20475
igraph Inparanoid8Db
1 268
list MSnSet
18 1
mzRident mzRpwiz
1 1
OrgDb Rle
2002 1852
SQLiteConnection TwoBitFile
1 5558
TxDb VcfFile
75 8
其中
FaFile就是fasta文件UCSC的bigwig格式存储了基因组信号值coverage情况,用每个坐标区间的测序深度来表示。它是wig格式的压缩后的带有索引的二进制格式[利用
wigToBigWig工具],速度更快。关于测序深度,可能我们对BAM文件比较熟悉,利用
samtools depth input.bam可以计算测序深度(基因组上每bp碱基覆盖到的reads数目),一般得到的结果文件是:第一列是染色体,第二列是比对到染色体上不同碱基的位置,第三列为覆盖第二列位点的reads数目。既然如此,为什么还要wig/bigwig或bedgraph文件呢?
因为人类基因组3G,BAM文件是非常大的,这种往往造成后续的可视化分析不好进行,比如一个3G左右的BAM文件,其包含的coverage信息在bigwig中只有十几M,一般ChIP-seq的覆盖度、测序深度分布在IGV的可视化中,使用bigwig比较频繁。这里只建立基础的认知,具体数据格式及可视化日后再来。
chainfile的作用就是进行基因组不同版本之间的坐标转换,如hg17/hg18/hg19/hg38OrgDb是做富集分析时主要用到的
BioStrings & BSgenome
这个包主要是用来处理基因序列的
准备基因组可以利用AnnotationHub或者BSgenome,其实AnnotationHub的基因组资源更多,但这里为了全面性考虑,主要介绍BSgenome的方法
BSgenome可以提供一个标准的注释信息,主要针对模式生物 其中关于人的,包括了hg17、18、19、38 小tip:
- 为什么看到下载的基因组中有N,还基本连续排列?表示没有测序出来的碱基,可以是ATCG的任意碱基
- 然后标准的CDS序列是ATCG四个大写字母
- 为什么还有小写的碱基?表示虽然测序成功但是序列复杂度比较低,一般是内含子(在成熟mRNA形成之前会被切除掉)或UTR(非编码)区域或者重复区域 =》 soft mask序列
- 最后,如果基因结构有不明白的,推荐看 下 https://vip.biotrainee.com/d/112--/4
BiocManager::install("BSgenome", version = "3.8")
library(BSgenome)
available.genomes() #总共91个,现在下载hg38
BiocManager::install("BSgenome.Hsapiens.UCSC.hg38", version = "3.8")
library("BSgenome.Hsapiens.UCSC.hg38")
# ?BSgenome.Hsapiens.UCSC.hg38
hg38 <- BSgenome.Hsapiens.UCSC.hg38
BSgenome的hg38基因组中都有什么信息?
可以看到物种、提供者、版本、发布日期、发布标准名称、染色体名称
染色体除了熟知的22对常染色体+X+Y+线粒体的,还有像chrUn_KI…这种,表示可以测出来确定是人的序列但拼不回去,还有像这种chr4_GL000008v2_random表示确定是人的4号染色体的序列但拼不回去。想看全部的名称使用 seqnames(hg38)
> hg38
Human genome:
# organism: Homo sapiens (Human)
# provider: UCSC
# provider version: hg38
# release date: Dec. 2013
# release name: Genome Reference Consortium GRCh38
# 455 sequences:
# chr1 chr2 chr3
# chr4 chr5 chr6
# chr7 chr8 chr9
# chr10 chr11 chr12
# chr13 chr14 chr15
# ... ... ...
# chrUn_KI270744v1 chrUn_KI270745v1 chrUn_KI270746v1
# chrUn_KI270747v1 chrUn_KI270748v1 chrUn_KI270749v1
# chrUn_KI270750v1 chrUn_KI270751v1 chrUn_KI270752v1
# chrUn_KI270753v1 chrUn_KI270754v1 chrUn_KI270755v1
# chrUn_KI270756v1 chrUn_KI270757v1
# (use 'seqnames()' to see all the sequence names, use the '$' or '[['
# operator to access a given sequence)
截取序列
seqlengths(hg38) # 获取上面seqnames各条染色体的长度
hg38$chr1 #或者 getSeq(hg38, "chr1")
# 可以像这样创建
str1 = getSeq(hg38, "chr1", start = 10001, end = 10030)
# 或者直接创建
# str1 = DNAString("TACCCTAACCCTAACTAACCCTAA")
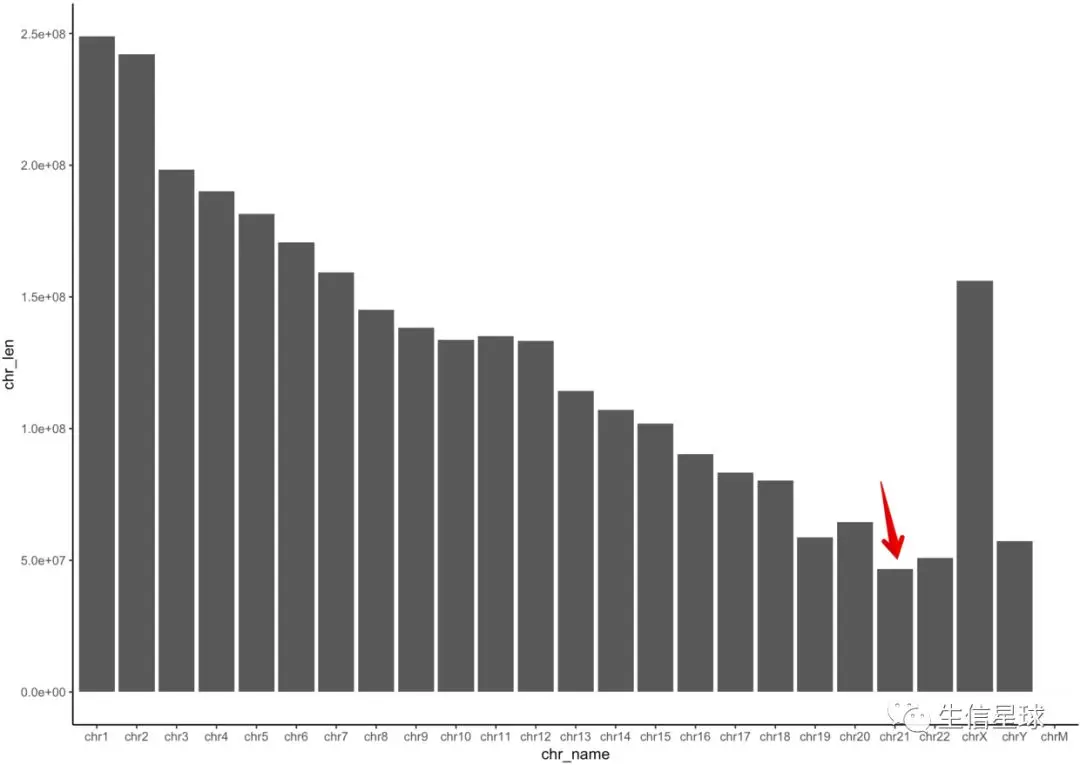
小操作:使用ggplot2对各条染色体长度进行可视化
# 使用ggplot2对各条染色体长度进行可视化
if(F){
hg38.len <- data.frame(chr_name = seqnames(hg38),
chr_len = as.numeric(seqlengths(hg38)))
hg38.len <- hg38.len[1:25,]
#为了避免按ASCII编码chr1、chr10、chr11...画图,我们先进行因子转化,保证画出来的是chr1、chr2、chr3...
hg38.len$chr_name <- factor( hg38.len$chr_name,
levels = as.character( hg38.len$chr_name))
library(ggplot2)
ggplot(hg38.len, aes(x = chr_name, y = chr_len)) +
geom_bar(stat = "identity")
}
可以非常直观看到:人类最小的染色体是21号
然后进行简单的探索
> reverse(str1) #反向序列
24-letter "DNAString" instance
seq: AATCCCAATCAATCCCAATCCCAT
> reverseComplement(str1) #反向互补序列
24-letter "DNAString" instance
seq: TTAGGGTTAGTTAGGGTTAGGGTA
> str1[1:15] #取其中一部分
15-letter "DNAString" instance
seq: TACCCTAACCCTAAC
> length(str1) #统计长度
[1] 24
> translate(str1) #CDS序列按照标准的密码子表翻译成氨基酸
8-letter "AAString" instance
seq: YPNPN*P* #其中*表示stop codon
> alphabetFrequency(str1) #统计str1中每个碱基出现的次数
A C G T M R W S Y K V H D B N - + .
9 10 0 5 0 0 0 0 0 0 0 0 0 0 0 0 0 0
可以看到上面统计str1中每个碱基出现的次数时除了常规的ATCGN,还有其他的一些字母,它们其实表示了
简并碱基,或者可以看看国际标准的兼并碱基
> IUPAC_CODE_MAP
A C G T M R W S Y K V
"A" "C" "G" "T" "AC" "AG" "AT" "CG" "CT" "GT" "ACG"
H D B N
"ACT" "AGT" "CGT" "ACGT"
统计GC信息:
> letterFrequency(str1,"GC") #统计GC信息
G|C
10
> letterFrequency(str1,"GC",as.prob = T) #统计GC信息(百分比)
G|C
0.4166667
> dinucleotideFrequency(str1,as.prob = T) #每个两两碱基组合百分比
AA AC AG AT CA CC CG
0.1739130 0.1739130 0.0000000 0.0000000 0.0000000 0.2608696 0.0000000
CT GA GC GG GT TA TC
0.1739130 0.0000000 0.0000000 0.0000000 0.0000000 0.2173913 0.0000000
TG TT
0.0000000 0.0000000
# trinucleotideFrequency(str1,as.prob = T) # 当然统计三个碱基组合也可以
取基因组指定染色体的一段序列
> str2 <- hg38[["chr1"]][10000:20000] #取基因组指定染色体的一段
> str2
10001-letter "DNAString" instance
seq: NTAACCCTAACCCTAACCCTAACCCTAACCCTAAC...AACTGAGACTGGGGAGGGACAAAGGCTGCTCTGT
# 然后可以像上面一样统计GC含量,比如统计chr2的GC含量
> letterFrequency(hg38[["chr2"]],"GC",as.prob = T)
G|C
0.3995527