012-重测序之BSA
刘小泽写于2018.7.16 表型与基因型相关联(基因定位)是遗传学上的重要问题。
什么是重测序Resequencing?
我们使用的基因组是怎么来的? 通过Denovo测序,也就是从头测序,拼接得到基因组
那么重测序呢? 已有的基因组发表出来以后,重新进行测序就不需要拼接,直接可以对测序reads进行短序列比对。简而言之就是对基因组序列已知的物种个体进行基因组测序。本质就是找变异。
通过这种方法,可以比较个体或者群体差异,建立该物种的变异数据库,挖掘重要性状的关键候选基因。利用重测序可以进行变异检测(SNP Calling)、遗传图谱构建、群体进化以及全基因组关联分析(GWAS)。
由于基因组的测序覆盖度不同,可分为全基因组重测序和简化基因组重测序。 **全基因组重测序(WGS)**是在全基因组水平上检测动植物重要性状相关的变异位点,可以检测SNP(单核苷酸多态性)、InDel(小片段插入缺失)、SV(结构变异)、CNV(拷贝数变异)、转座子变异等。
利用鸟枪法建立350bp文库,测序深度10-30X,适用于有参物种,成本较高 (SNP/InDel >10X; SV > 20X; CNV > 30X, 转座子> 20X)增加测序深度可以提高基因组覆盖度和变异检出率
简化基因组测序(GBS, Genotyping by Sequencing) 将基因组DNA进行酶切,然后对酶切片段序列进行测序,一般能捕获全基因组的1%序列,主要用于检测SNP,成本低适合大规模样品标记筛选。
什么是BSA?
BSA是一种检测位于特定染色体片段上标记的方法,又叫Mapping-by-sequencing。理论上,任何一对具有相对形状的亲本杂交产生的分离后代都适用,也就是针对质量性状单基因或数量性状主效基因的定位。但BSA一次只能分析一个性状
建库时两个亲本分别建库,子代分别混池建库,共四个,文库片段大小350bp 测序深度一般亲本>10X,子代混池>20X
一般要经过以下几步:
- 发现相对性状:可能是人为诱变,可能是自然发现
- 构建群体: 异交(outcross)或回交(backcross)
- 选取目标性状群体
- 测序分析
如何分析?
拿回来数据,第一步总是要对数据过滤清洗,去接头去不合格reads;第二步是序列比对,常用bwa、bowtie2;第三步SNP calling, GATK(最全最好用的)、samtools+bcftools(易上手的);第四步作图确定可能区域;第五步变异位点注释
使用的软件:
主要是
SHOREmap 、samtools、bcftools、fastqc、multiqc、bwa,可以先把软件安装好,最快的方法是conda install -p /your conda path/ tools 。
下载测试数据:
这一次分析拟南芥回交BC群体数据以及参考基因组、注释文件
#sample data download in dir data 下载到data下
mkdir -p ~/bwa/data && cd ~/bwa/data
axel http://bioinfo.mpipz.mpg.de/shoremap/data/software/BC.fg.reads1.fq.gz
axel http://bioinfo.mpipz.mpg.de/shoremap/data/software/BC.fg.reads2.fq.gz
axel http://bioinfo.mpipz.mpg.de/shoremap/data/software/BC.bg.reads1.fq.gz
axel http://bioinfo.mpipz.mpg.de/shoremap/data/software/BC.bg.reads2.fq.gz
# reference genome & gff in dir reference 下载到reference下
mkdir -p ~/bwa/reference && cd ~/bwa/reference
wget -c https://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_chromosome_files/TAIR10_chr_all.fas
wget -c https://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff
#chr_sizes
wget http://bioinfo.mpipz.mpg.de/shoremap/data/software/chrSizes.txt
分析过程:
质控
#进入到数据文件夹,例如data下
unzip *.gz ls *.fq | xargs -i fastqc {} multiqc .
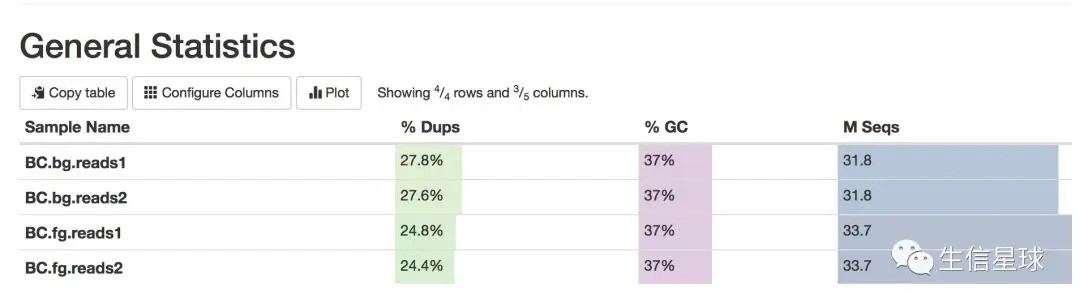
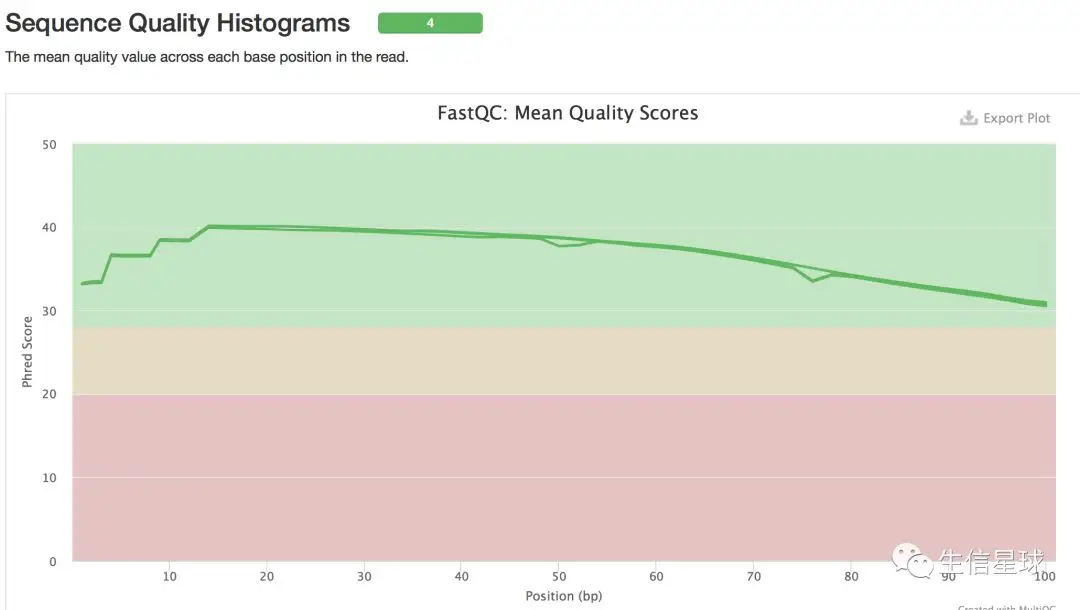
这应该是清洗以后的数据了,质量还不错。如果自己拿回来的数据,有时需要去接头,去掉前几bp的低质量序列
序列比对
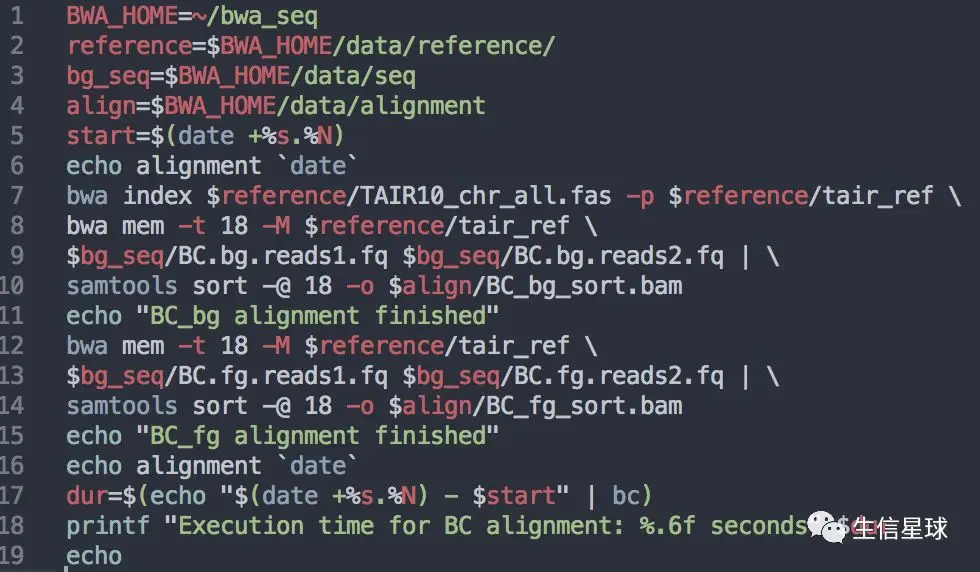
重测序中的比对使用最多就是bwa,分为两步: 先对参考基因组构建索引index,然后使用bwa mem比对;【当然构建索引使用bowtie2软件的bowtie-build命令也是可以的】 比对的结果是sam格式,我们这里用samtools sort一步将管道输入给它的sam文件排序并转换格式为bam文件 【你可能注意到还有一些start、end、echo之类的命令,他们是为了提示用户进行到了哪一步,并且计算运行时间;这些都是可有可无的,属于点缀】
SNP calling 这是基于贝叶斯定理,你肯定眼熟~后期讲统计概率先讲它!太重要了!
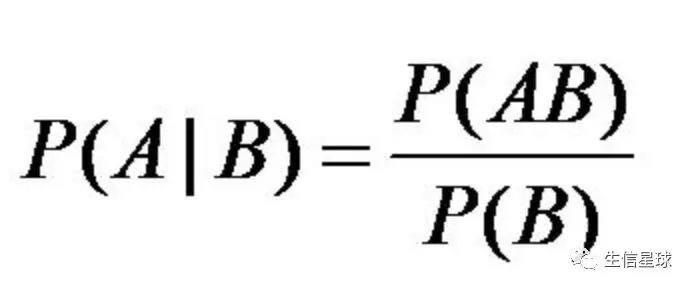 假如我们之前比对的短序列和参考基因组有差异了,那么还不能确定他就是一个变异,作为候选人,需要接受多方考证:
一个是测序准确度,测不准也有可能会有变异;
一个是比对软件的精度,都是人开发的,难免有误差:
一个是抽样的误差,就是群体选材时产生的;
如何确定它是不是一个真实的变异呢?这里贝叶斯定理就派上了大用场~
假如我们之前比对的短序列和参考基因组有差异了,那么还不能确定他就是一个变异,作为候选人,需要接受多方考证:
一个是测序准确度,测不准也有可能会有变异;
一个是比对软件的精度,都是人开发的,难免有误差:
一个是抽样的误差,就是群体选材时产生的;
如何确定它是不是一个真实的变异呢?这里贝叶斯定理就派上了大用场~比如基因组上一个位点有50条reads比对上,但是其中的40条都与参考基因组不同,那么很有可能他是一个真的变异位点,至于什么变异下面再分析
这里用的samtools是最新版本1.8,mplieup主要功能主要是生成BCF、VCF文件。这里-v指定输出为vcf文件,否则默认生成pileup格式文件;-f指定构建过索引的参考序列;-u是uncompressed,输出未压缩的VCF文件;

SHOREmap确定候选区域
SHOREmap的安装还是比较复杂的,需要有root权限去安装两个动态库 首先,确定自己的系统是不是64位
uname -m显示X86_64就是64位; 其次,安装依赖的库 DISLIN ,而doslin自己又需要两个动态库#检查安装doslin的两个动态库 #检查libXm.so*是否安装,在命令行输入 ls /usr/lib/libXm.so* #如果显示'ls: cannot access /usr/lib/libXm.so*: No such file or directory‘,就需要安装【接下来需要root权限】 sudo apt-get update sudo apt-get install libmotif4 #再检查一遍 ls /usr/lib/libXm.so* #检查libXt.so是否安装 ls /usr/lib/x86_64-linux-gnu/libXt.so #32位系统输入:ls /usr/lib/i386-linux-gnu/libXt.so #出现'ls: cannot access /usr/lib/x86_64-linux-gnu/libXt.so: No such file or directory‘,就需要安装 sudo apt-get install libxt-dev #再检查一遍#安装完doslin需要的动态库后,进入软件目录,开始下载doslin cd /path/to/software # 下载 wget ftp://ftp.gwdg.de/pub/grafik/dislin/linux/i586_64/dislin-11.1.linux.i586_64.tar.gze # 解压缩 tar -zxvf dislin-11.0.linux.i586_64.tar.gz cd dislin-11.0 # 加入系统路径 mkdir -p $HOME/biosoft/dislin DISLIN=$HOME/biosoft/dislin export DISLIN # 开始安装 ./INSTALL # 复制dislin_d.h 到dislin文件夹中 cp ./examples/dislin_d.h $DISLIN然后才开始正题——安装SHOREmap,当前最新版本是3.6版本
#里面的安装路径需要个性化自己定制哦 cd /your software path/ wget http://bioinfo.mpipz.mpg.de/shoremap/SHOREmap_v3.6.tar.gz # 替换SHOREmap下的dislin的一些文件 tar -zxvf SHOREmap_v3,6 rm dislin/*dislin_d.* cp $DISLIN/*dislin_d.* dislin # 编辑/etc/profile[root用户]或.bashrc[非root] vi ~/.bashrc export LD_LIBRARY_PATH=/path/to/SHOREmap_v3.6/dislin # 退出保存.bashrc source ~/.bashrc # 到之前安装的文件夹下 cd & cd src/SHOREmap_v3.6 #进行编译(可选,如果没有g++) #sudo apt-get install build-essential make #【编译过程如果出错,有可能动态库文件路径设置不正确,把它放到/etc/profile或者/.bashrc的上面】 # 将编译文件拷贝到习惯的文件夹中,然后添加执行路径 cp SHOREmap ../../biosoft/SHOREmap_v3.6 echo "export $HOME/bisoft/SHOREmap_v3.6" >> ~/.bashrc source ~/.bashrc # 检查是否安装成功 SHOREmap
以下是官网给出的针对Outcross和Backcross群体的不同分析流程,但是都要求先转换格式【我们这里的测试流程是针对的Backcross】
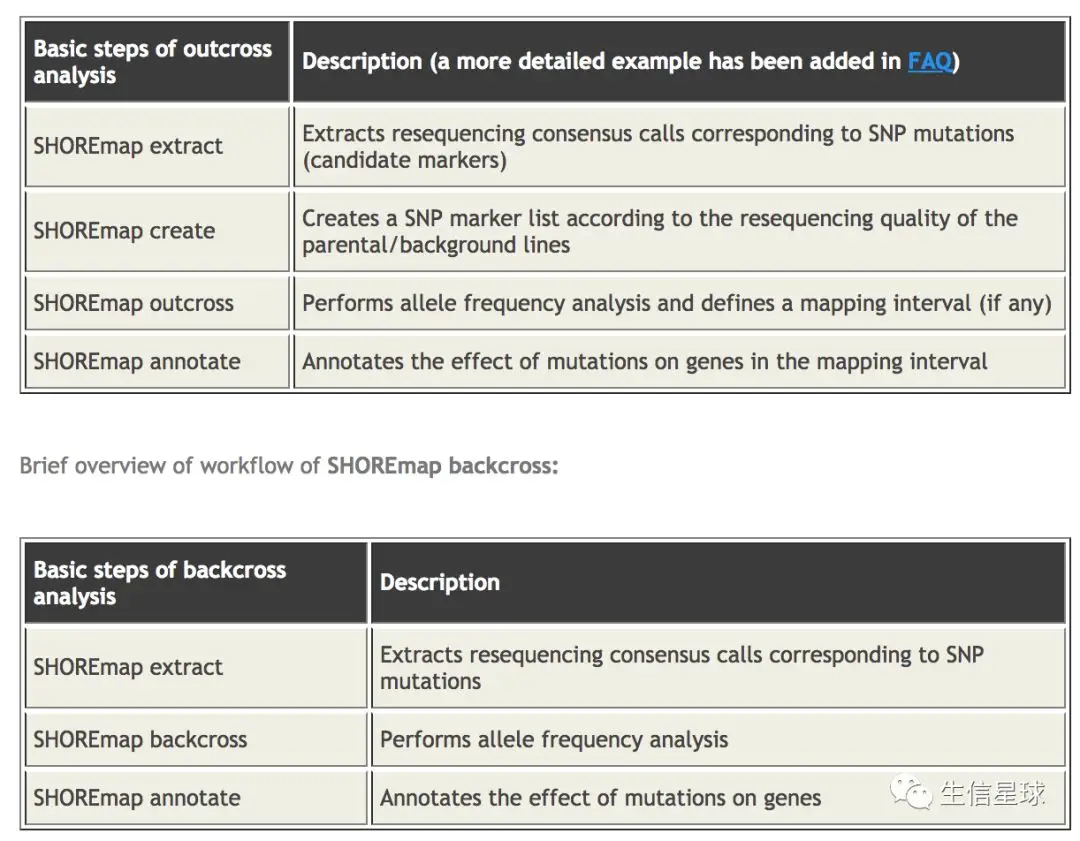
转换格式:将vcf转换成软件能认识的格式 基本两个参数就是
--marker和--folder【注意:因为这里是用的两台服务器,前面跑的那台没有root权限,又注册了一个腾讯云服务器,获得了权限后安装的各种库,接下来的分析也用的是云服务器,因此代码可能和上面有出入】SHOREmap convert --marker ./variant/BC_bg_raw_variant.vcf --folder background/ SHOREmap convert --marker ./variant/BC_fg_raw_variant.vcf --folder foreground/ #生成三个文件👇
提取候选分子标记的一致性信息:SHOREmap extract
四个参数
--chrsizes、--folder、--marker、--consen分别对应染色体长度信息、指定输出目录、上面转换后的#background SHOREmap extract --chrsizes chrSizes.txt --folder ../background/ --marker ../background/1_converted_variant.txt --consen ../background/1_converted_consen.txt -verbose #foreground SHOREmap extract --chrsizes chrSizes.txt --folder ../foreground/ --marker ../foreground/1_converted_variant.txt --consen ../foreground/1_converted_consen.txt -verbose #生成 extracted_consensus_0.txt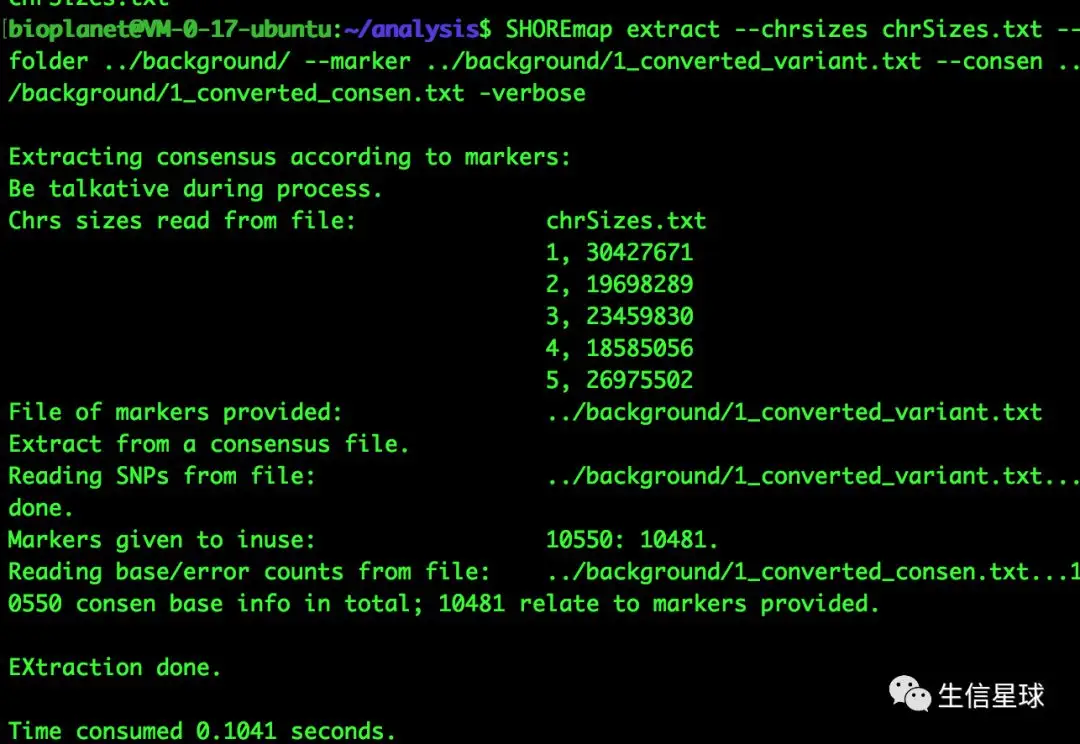
回交分析SHOREmap backcross 用来分析回交作图群体得到的重组后代混池数据,想过滤出所有参考基因组和测序池之间的不同部分用于找到突变点特异部分。如何确保不是自然变异或者是测序错误而产生的特异部分呢?要求测序池中选择的部分要多次出现在亲本或背景中。再根据前景/背景的碱基识别,base calls,质量/覆盖率/等位基因等信息,确定是否把保留的SNP位点作为分子标记
必选的参数有三个:
染色体大小文件
--chrsizes,包括两行,一是染色体位置,二是大小;--marker通过SHOREmap convert生成的候选marker文件 converted_variant.txt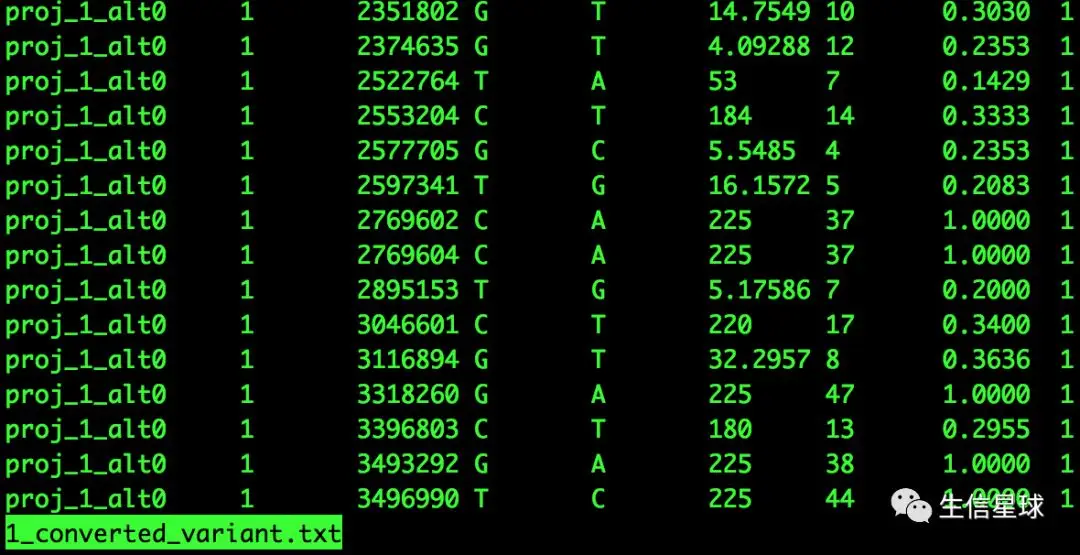
这几列分别代表:项目名称、染色体ID、SNP标记位点位置、参考序列上的碱基、变异的碱基、可变碱基质量(0-40)【也是用Phred换算】、符合这种碱基改变的reads数、前一项占总覆盖度的百分比、覆盖到SNP位置的比对平均命中数(也就是这个区间能检测到SNP的位点数)
--folder输出文件夹
SHOREmap backcross --chrsizes chrSizes.txt --marker ../foreground/1_converted_variant.txt --consen extracted_consensus_0.txt --folder ./ -plot-bc --marker-score 40 --marker-freq 0.0 --min-coverage 10 --max-coverage 80 -bg ../background/1_converted_variant.txt --bg-cov 1 --bg-freq 0.0 --bg-score 1 --non-EMS --cluster 1 --marker-hit 1 -verbose
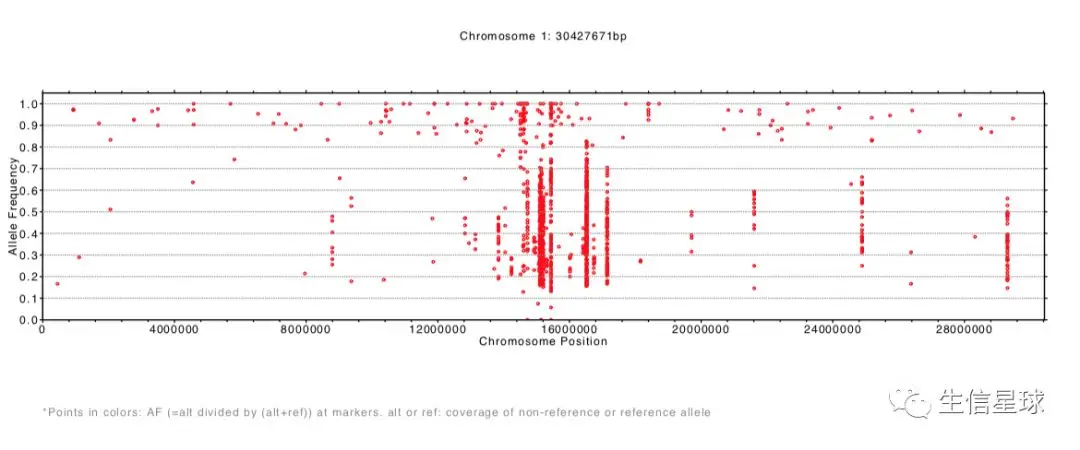
显示的结果及文件:

结果生成了每一条染色体等位基因频率分布pdf文件
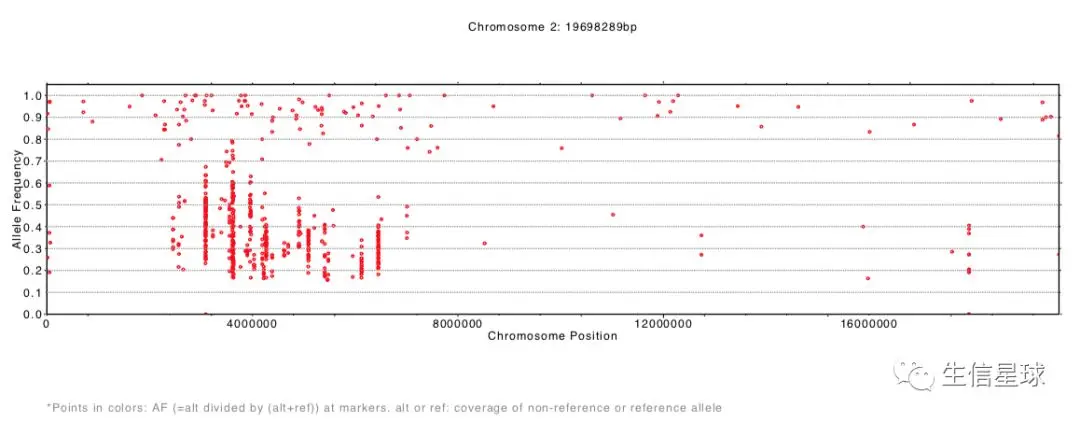

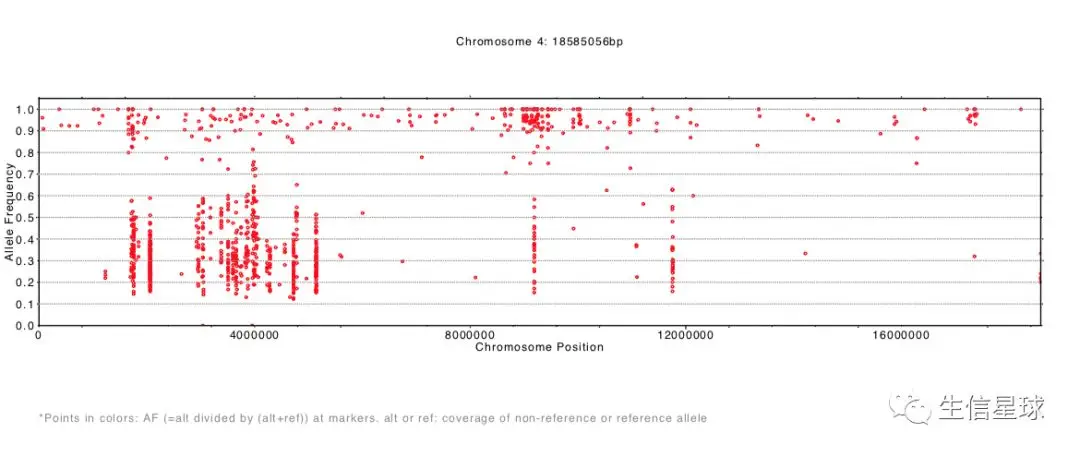
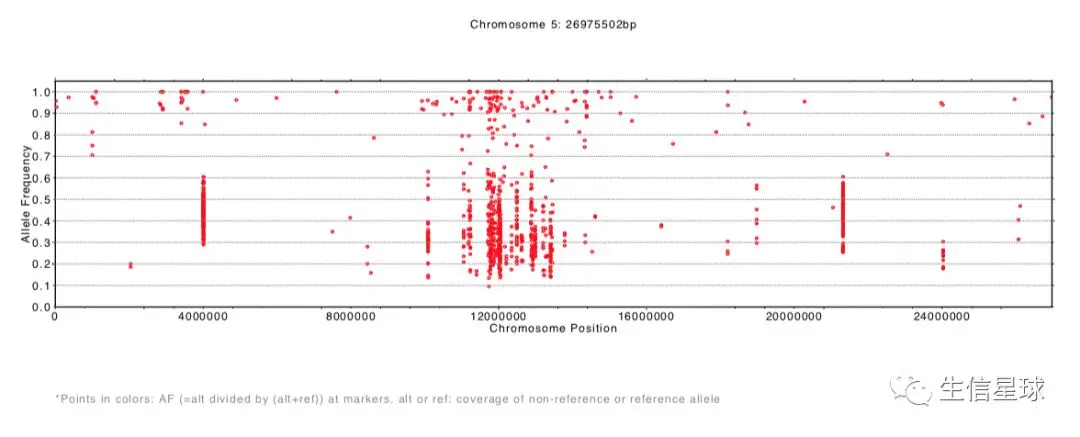
SHOREmap annotate:注释候选区域
SHOREmap annotate --chrsizes chromsize.txt --folder ../analysis/ --snp ../foreground/1_converted_variant.txt --chrom Chr3 --start 1 --end 4000000 --genome ../genome/TAIR10_chr_all.fas -gff../annotation/TAIR10_GFF3_genes.gff